Corresponding author: Shinji Kume, skume@belle.shiga-med.ac.jp
DOI: 10.31662/jmaj.2020-0005
Received: January 21, 2020
Accepted: March 9, 2020
Advance Publication: April 7, 2020
Published: April 15, 2020
Cite this article as:
Kume S, Maegawa H. Lipotoxicity, Nutrient-Sensing Signals, and Autophagy in Diabetic Nephropathy. JMA J. 2020;3(2):87-94.
Diabetic nephropathy is a leading cause of proteinuria, kidney fibrosis, and subsequent end-stage renal disease. The renal prognosis of diabetic patients with refractory proteinuria is extremely poor. Therefore, identification of novel therapeutic targets to combat this serious condition and improve renal prognosis is urgently necessary. In diabetic patients, in addition to blood glucose levels, serum levels of free fatty acids (FFAs) are chronically elevated, even during postprandial periods. Of the various types of FFAs, saturated FFAs are highly cytotoxic and their levels are elevated in the serum of patients with diabetes. Thus, an increase in saturated FFAs is currently thought to contribute to proximal tubular cell damage and podocyte injury in diabetic nephropathy. Therefore, protecting both types of kidney cells from saturated FFA-related lipotoxicity may become a novel therapeutic approach for diabetic patients with refractory proteinuria. Interestingly, accumulating evidence suggests that controlling intracellular nutrient signals and autophagy can ameliorate the FFA-related kidney damage. Here, we review the evidence indicating possible mechanisms underlying cell injury caused by saturated FFAs and cell protective roles of intracellular nutrient signals and autophagy in diabetic nephropathy.
Key words: Free fatty acid, proximal tubular cell, podocyte, lipotoxicity, proteinuria, autophagy
Diabetic nephropathy is a world’s leading cause of end-stage renal disease, and its prevalence is increasing. Thus, to improve healthy lifespan in patients with diabetic nephropathy, a complete understanding of its pathogenesis and effective therapeutic strategies are required. Glomerulosclerosis and albuminuria are characteristic histological and clinical features of diabetic nephropathy. Recent clinical studies have demonstrated that intensive care treatment including strict glycemic controls and use of a renin-angiotensin system (RAS) antagonist can abolish or diminish low-grade proteinuria, such as microalbuminuria, in early-stage diabetic nephropathy(1), (2), (3), (4). However, once proteinuria develops to massive and refractory proteinuria, the resultant tubulointerstitial damage leads to nephron loss and subsequent renal dysfunction. To better improve renal outcome in diabetic nephropathy, it is therefore necessary to identify new therapeutic targets to prevent stage progression from low-grade to massive and refractory proteinuria or to protect proximal tubular cells from proteinuria-related toxicity.
In addition to the above-mentioned current standard of approach to diabetic nephropathy, some emerging therapeutic agents have been becoming to be applied in clinical settings. Several anti-diabetic agents currently used in clinical settings may have some pleiotropic renoprotective effects. For instance, thiazolidinediones have direct renoprotective action experimental models (5), and their anti-albuminuric effect was observed in a meta-analysis (6). Inhibitors of dipeptidyl peptidase-4 (DPP-4) have emerged in the treatment paradigm of diabetes. Some experimental models have indicated possible renoprotective benefits of DPP-4 inhibitors (7), and actually, a clinical trial suggested anti-albuminuric effect of this type agent in type 2 diabetic patients treated with RAS inhibitors (8). Furthermore, a recent clinical trial using a sodium-glucose co-transporter 2 (SGLT2) inhibitor, empagliflozin, appears to improve the composite hard renal endpoints in type 2 diabetic patients at high cardiovascular risk (9), in which some pleiotropic effects such as persistent hyperketonemia ketone bodies and/or hematocrit are suggested to be involved in the renoprotective mechanism of the SGLT2 inhibitor (10). In addition, other drugs, which have been invested to ameliorate directly pathogenic responses such as inflammation, oxidative stress, and vasoconstriction, have recently been developed and expected as novel drugs for diabetic nephropathy. These drugs include selective C-C chemokine receptor type 2 antagonist (11), vitamin D receptor activators (12), nuclear respiratory factor 2 activator, bardoxolone methyl (13), and selective endothelin-A antagonism (14). Thus, aside from the current standard therapy, some other therapeutic options have been expected to improve renal outcome in diabetic nephropathy in the future. However, until their long-term safety and efficacy are ensured, their therapeutic effectiveness is still uncertain. Thus, we still need to identify a novel therapeutic target to improve renal outcome in diabetic nephropathy.
Insulin has a variety of biological effects. One of its important physiological roles is to facilitate glucose uptake into peripheral tissues and prevent the release of free fatty acids (FFAs) from adipose tissue during postprandial periods (15). In diabetes, reduced insulin action on skeletal muscle, the liver, and adipose tissue causes hyperglycemia and impairs the normally rapid postprandial decline of serum FFA levels. Thus, in addition to hyperglycemia, high FFA levels caused by insufficient insulin action represent a potential pathogenic factor in diabetic complications. Indeed, increasing experimental evidence supports a pathogenic role for FFAs in the progression of β cell dysfunction-related diabetic complications in late-stage diabetes (16), (17). Furthermore, experimental evidence has accumulated demonstrating a pathogenic role of FFAs, especially saturated FFAs, in podocyte damage, leading to massive proteinuria and proteinuria-related tubular cell damage in diabetic nephropathy. Thus, reducing saturated FFA-mediated cell toxicity may serve as an emerging therapy for refractory diabetic nephropathy. However, no drug and strategy aimed at ameliorating saturated FFA-mediated cell toxicity have not been developed in clinical settings.
In this review, we describe our reasons for focusing on FFA levels as a pathogenic factor in diabetic nephropathy and also discuss mechanisms underlying cell injury by saturated FFAs in this serious diabetic complication, which may help contribute to future development of novel drugs for diabetic nephropathy.
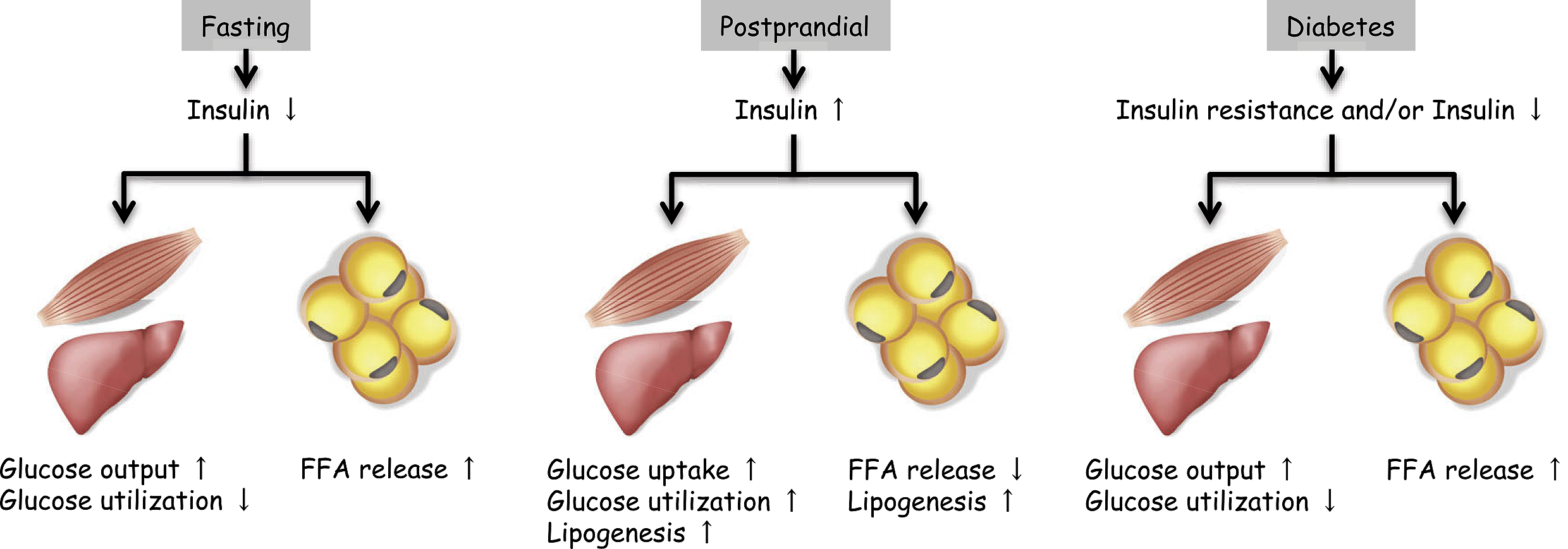
Insulin action on peripheral metabolic tissues decreases as a result of deficient insulin secretion in type 1 diabetes or insulin resistance combined with insufficient insulin secretion in type 2 diabetes. The physiological role of insulin in skeletal muscle and the liver is to facilitate glucose uptake and utilization during the postprandial period (Figure 1). Thus, impaired insulin action in these tissues causes the hyperglycemia that occurs in diabetes. Another important physiological action of insulin is its enhancement of lipogenesis and inhibition of FFA released by adipose tissue (Figure 1). Because FFAs are used as an energy source in various tissues during fasting, fasting serum FFA levels are very high in both diabetic and non-diabetic subjects. In non-diabetic subjects, serum FFA levels rapidly decrease during the postprandial period in an insulin-dependent manner. In contrast, in diabetic patients, who have impaired insulin action in adipose tissue, high FFA levels are sustained even during the postprandial period (Figure 1). Thus, in addition to hyperglycemia, high postprandial FFA levels may represent a typical metabolic alteration in diabetes. The risk of hyperglycemia in diabetic nephropathy has been well documented; it is now necessary to focus more on high FFA levels, also caused by insufficient insulin action, as a risk factor for diabetic nephropathy.
FFAs are classified into saturated, monounsaturated, and polyunsaturated fatty acids. These FFA classes have specific roles in metabolism and in the pathogenesis of diabetes-related diseases. Previous reports have demonstrated that diabetic patients exhibit abnormal fractions of serum FFAs, with proportionately higher levels of saturated FFAs compared with other types (18), (19).
A number of previous studies showed that saturated FFAs, such as palmitate and steric acid, induced cell damage and dysfunction in various tissues and that such damage was ameliorated by unsaturated FFAs, such as oleate (a monounsaturated fatty acid) and ω-3 polyunsaturated fatty acid (20), (21). These observations suggest involvement of an imbalance between FFA classes in the pathogenesis of diabetic complications and indicate the possibility that control of FFA type, as well as quantity, might represent a novel therapeutic approach for diabetic nephropathy.
Proteinuria is a major symptom of diabetic nephropathy and indicates impairment of the glomerular filtration barrier. Albumin, a major excreted macromolecule in proteinuria, is a carrier protein that binds to a number of molecules in the blood. Because serum FFAs bind to albumin, they are filtered through the impaired glomerular filtration barrier and reabsorbed by proximal tubular cells, along with albumin, in patients with refractory proteinuria. These albumin-bound FFAs then induce oxidative stress and inflammatory cytokine production in the proximal tubular cells, leading to tubulointerstitial damage and subsequent renal dysfunction (22), (23), (24), (25). Thus, protecting the proximal tubular cells from FFA-induced lipotoxicity is a potential therapeutic approach for improving the prognosis for renal function in diabetic patients with refractory proteinuria.
A pathogenic role of FFAs bound to albumin in the development of proximal tubular cell damage has been demonstrated by experimental studies in humans and animals. One such study showed that severe tubulointerstitial damage was induced by FFA-bound albumin, but not by FFA-free albumin, in a mouse experimental model (26). Furthermore, a human cohort study showed that diabetic patients with proteinuria in the nephrotic range had higher urinary levels of FFAs and more severe tubulointerstitial injury than patients with non-diabetic minimal-change nephrotic syndrome (27). A more recent paper describing a human cohort study using RNA microarrays demonstrated that lipid metabolism was altered in proximal tubular cells of patients with kidney diseases, in a manner strongly associated with the progression of their tubulointerstitial lesions (28). Therefore, chronic elevation of FFA levels is implicated in the proximal tubular cell damage caused by proteinuria in diabetic nephropathy.
We propose several molecular mechanisms that could underlie saturated FFA-mediated proximal tubular injury (Figure 2).
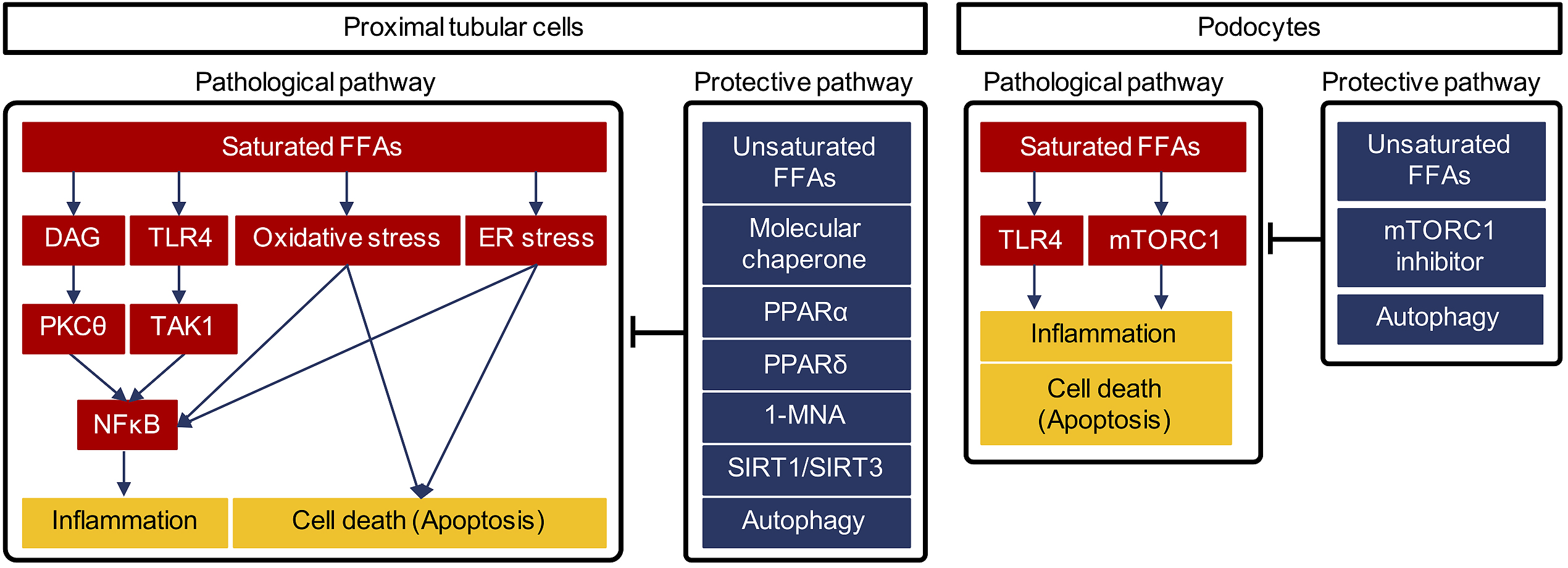
Diacylglycerol (DAG) is a well-known pathogenic factor in diabetic complications. Hyperglycemia increases DAG formation, leading to hyper-activation of protein kinase C isoforms (PKCs) (29). Activation of PKC signaling is strongly associated with fibrosis, cell death, and inflammation, and its inhibition is considered a potential therapeutic approach to diabetic nephropathy (30). Our recent study showed that palmitate, a saturated FFA, led to increased intracellular DAG accumulation and subsequent activation of PKCθ in proximal tubular cells (20). Saturated FFA-mediated activation of the DAG-PKCθ pathway induced an inflammatory response via upregulation of NFκB and mitochondrial cell death in cultured proximal tubular cells (20). Thus, high levels of saturated FFAs may also contribute to activation of DAG-PKC pathways in human diabetic nephropathy, even though such activation has long been regarded as being mediated primarily by hyperglycemia.
Toll-like receptor 4 (TLR4) is implicated in the recognition of various bacterial wall components (31). Activation of toll-like receptor-mediated signaling can induce inflammation. Recent reports showed that saturated FFAs can act as endogenous ligands for TLR4, which, when activated, induces inflammation and contributes to development of several diseases (32), (33). Proximal tubular cells express TLR4 along the brush border (34). We previously reported that palmitate activated TLR4 signaling and induced inflammation via activation of the signaling pathway downstream of TLR4 in proximal tubular cells (35). Interestingly, peroxisome proliferator-activated receptor (PPAR) δ agonists exerted anti-inflammatory effects via inactivation of transforming growth factor β-activated kinase (TAK1), downstream of TLR4. TAK1 inactivation inhibited NFκB activation by enhancing degradation of IκB, an NFκB suppressor (35). A PPAR δ agonist, GW501516, inhibited NFκB-mediated overexpression of MCP-1 by directly inhibiting TAK1 activity (35). Thus, although PPARδ agonists were originally developed as activators of mitochondria function and fatty acid oxidation, they are likely to have a pleiotropic effect as a direct inhibitor of TLR4-mediated inflammation. PPARδ agonists may therefore represent novel potential therapeutic agents for protection of proximal tubular cells from FFA-related lipotoxicity.
Fenofibrate is a PPARα agonist used clinically for treating hypertriglycemia. Experimental evidence demonstrated that this agent ameliorated proximal tubular injury induced by palmiate (23) and that PPARα deficiency in mice exacerbated kidney injury induced by FFA-bound albuminuria (36). In contrast to the renoprotective mechanism of PPARδ agonists, PPARα inhibited palmitate-induced proximal tubular cell injury by enhancing fatty acid oxidation and subsequently reducing saturated FFA-related lipotoxicity in both mice and cultured proximal tubular cells (23). A clinical study, the Fenofibrate Intervention and Event Lowering in Diabetes study, showed that the PPARα agonist fenofibrate prevented initiation and progression of albuminuria in patients with type 2 diabetes (37), (38). Unfortunately, fenofibrate cannot be given to patients with renal dysfunction because it is renally excreted. However, for patients with normal renal function, PPARα agonists such as fenofibrate might be useful for treating early diabetic nephropathy.
Endoplasmic reticulum (ER) stress is also recognized as a pathogenic factor in diabetic complications (39). One study found that palmitate stimulation induced excessive ER stress and activation of C/EBP homologous protein, leading to cell death, in cultured proximal tubular cells (40). Supplementation with molecular chaperones to increase protein folding capacity in the ER reduced this palmitate-induced proximal tubular cell damage (40). These findings indicate that enhancing ER capacity by molecular chaperone supplementation might be a therapeutic approach for diabetic nephropathy.
Nicotinamide adenine dinucleotide (NAD) metabolism has recently been considered as a potential therapeutic target for several diseases, including diabetes and age-related neurodegenerative disorders (41), (42), (43). Our recent work showed that palmitate increased expression of nicotinamide n-methyltransferase, an NAD metabolizing enzyme (44). The product of nicotinamide n-methyltransferase activity, 1-methylnicotinamide, reduced mitochondrial oxidative stress and cell death in FFA-induced renal tubular damage (44). 1-methylnicotinamide may, therefore, serve as a treatment for lipotoxicity-mediated renal injury in diabetic patients with refractory proteinuria.
Calorie restriction extends lifespan in all organisms and is renoprotective. It also has anti-obesity and anti-diabetic effects in mammals (45). Studies designed to identify factors responsible for life extension by calorie restriction have identified a number of potential anti-aging molecules. Of these, sirtuins, which are known to induce autophagy, are now the focus of approaches to prolonging healthy lifespan (46), (47). Interestingly, several recent studies, including ours, showed that sirtuin activity and the process of autophagy are both crucial for renoprotection in diabetic nephropathy.
Sirtuins are NAD-dependent deacetylases and are classified into seven types in mammals (46). These enzymes sense changes in intracellular NAD concentrations and control cellular activity in response to intracellular energy state. Among these enzymes, SIRT1 has been suggested as a potential therapeutic target in several conditions, such as diabetes and kidney disease (48), (49), (50). Indeed, one study showed that SIRT1 overexpression, specifically in proximal tubular cells, ameliorated progression of diabetic nephropathy in mice (51). Furthermore, some SIRT1 activators, including resveratrol, have been shown to inhibit development of diabetic nephropathy in mice (52). Thus, SIRT1 activation is currently under investigation as a therapeutic approach for various diseases, including diabetes and its vascular complications.
In addition to SIRT1, SIRT3, which is localized in mitochondria, is associated with alleviating lipotoxicity-mediated tubular injury. Interestingly, SIRT3 expression levels have been shown to decrease in mouse kidneys damaged by FFA-bound proteinuria and in cultured proximal tubular cells treated with palmitate (22). Furthermore, decreased SIRT3 expression level was associated with increased MCP-1 expression in the kidneys damaged by FFA-bound proteinuria, whereas overexpression of SIRT3 reduced reactive oxygen species accumulation by enhancing mitochondrial oxidative capacity in proximal tubular cells exposed to palmitate (22). Thus, decreased SIRT3 expression may be involved in the pathogenesis of lipotoxicity-mediated tubular cell damage, and activation of SIRT3 may be another potential therapeutic target, although no specific SIRT3 activator is currently available.
Autophagy is an intracellular degradation system that maintains intracellular homeostasis by removing damaged proteins and organelles during stress conditions, including starvation, hypoxia, and ER stress (53). Autophagy machinery is needed across species to overcome starvation (54), and its alteration is involved in the pathogenesis of many diseases including diabetes and neurodegenerative diseases (53). Interestingly, there is increasing evidence that autophagy exerts a renoprotective effect during various forms of renal toxic stress, aging, ischemia, and treatment with anti-cancer drugs (55). We recently reported that autophagy activity was suppressed in the kidneys of mice with high-fat diet (HFD)-induced obesity and type 2 diabetes, although the detailed factor to inhibit the activity in the kidney (56). More importantly, suppression of autophagy exacerbated proteinuria-related tubular cell damage in obese, type 2 diabetic, proximal tubular cell-specific autophagy-deficient mice (56). HFD feeding is a standard model for insulin resistance (56). Therefore, autophagy insufficiency under conditions of insulin resistance, such as in type 2 diabetes, may be a crucial molecular mechanism underlying lipotoxicity-mediated proximal tubular cell injury. Furthermore, autophagy activators may therefore be possible treatments for diabetic nephropathy.
Proteinuria is caused by disruption to the glomerular filtration barrier that is maintained by the podocyte. In recent reports, podocytes were shown to be damaged by saturated FFAs, via similar molecular mechanisms as proximal tubular cells, in diabetic nephropathy. Thus, saturated FFA-related lipotoxicity is likely to be associated with initial proteinuria pathogenesis, as well as with the tubulointerstitial lesions associated with proteinuria in diabetic nephropathy.
As mentioned previously, saturated FFAs have been shown to activate TLR4 and induce inflammation in proximal tubular cells. In cultured podocytes, high glucose with saturated FFAs has been shown to increase TLR4 expression and proinflammatory cytokine synthesis (57). Inhibition of the TLR4 signaling pathway has been demonstrated to abolish saturated FFA-induced proinflammatory cytokine synthesis, which indicates a potential renoprotective role for TLR4 inhibition in diabetic podocytopathy. C/EBP homologous protein is a marker of activated ER stress and induces ER stress-related apoptosis. Expression of this protein and subsequent apoptosis were increased in cultured podocytes treated with saturated FFAs, and these effects were attenuated by co-treatment with unsaturated fatty acids (58). These pathogenic mechanisms are very similar to those demonstrated in the proximal tubular cell study.
In addition, we recently proposed a renoprotective mechanism associated with calorie restriction in another podocyte study. The mammalian target of rapamycin (mTOR) complex 1 (mTORC1) is a nutrient-sensing kinase that regulates a wide range of cellular processes and can sense hyper-nutrient states such as hyperglycemia and amino acid sufficiency (59). Although mTORC1 is physiologically essential for cell growth, differentiation, protein translation, and inhibition of autophagy, its hyper-activation was recently reported to be strongly associated with pathogenesis of diabetic podocytopathy (60), (61), (62). Thus, revealing the molecular mechanisms underlying diabetes-related mTORC1 activation may inform development of new therapies for diabetic nephropathy. Our recent study showed that palmitate activated mTORC1, leading to podocyte apoptosis and that this was antagonized by oleate, a monounsaturated FFA (21). Thus, imbalances between FFA classes in diabetic patients may cause diabetes-related hyper-activation of mTORC1 in podocytes and subsequent cell damage.
Moreover, podocyte-specific autophagy-deficient mice developed podocyte loss and massive proteinuria in an HFD-induced diabetic model for inducing minimal proteinuria (63). Because mTORC1 is a potent inhibitor of autophagy, autophagy insufficiency may be involved in the mTORC1-dependent podocytopathy that occurs in diabetes. Thus, similar to the results in proximal tubular cells, altered intracellular nutrient-sensing signals and autophagy are likely to be involved in diabetic podocytopathy (64), (65). These findings may contribute to the development of new therapeutic strategies for diabetic podocytopathy.
We have reviewed the mechanisms underlying FFA-mediated damage in proximal tubular cells and podocytes. This overall injury process may provide novel therapeutic targets to improve renal outcome in diabetic patients with refractory proteinuria. Although several molecular mechanisms of saturated FFA-mediated kidney injury have recently been revealed, there is currently no treatment available. Thus, further basic and clinical research in this field is needed. Our review should provide useful knowledge and perspectives for future studies on the relationship between FFA-related lipotoxicity and the pathogenesis of diabetic nephropathy.
This article is based on the study, which received the Medical Research Encouragement Prize of The Japan Medical Association in 2019.
None
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science grant number 25713033 to S. K.
The author thanks all members of the Department of Medicine, Shiga University of Medical Science, for their supports.
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837-53.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977-86.
Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract. 1995;28(2):103-17.
Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861-9.
Isshiki K, Haneda M, Koya D, et al. Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes. 2000;49(6):1022-32.
Sarafidis PA, Stafylas PC, Georgianos PI, et al. Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am J Kidney Dis. 2010;55(5):835-47.
Kanasaki K, Shi S, Kanasaki M, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63(6):2120-31.
Groop PH, Cooper ME, Perkovic V, et al. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care. 2013;36(11):3460-8.
Wanner C, Inzucchi SE, Lachin JM, et al. EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375(4):323-34.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diabetes Care. 2016;39(7):1108-14.
de Zeeuw D, Bekker P, Henkel E, et al. CCX140-B Diabetic Nephropathy Study Group. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. 2015;3(9):687-96.
de Zeeuw D, Agarwal R, Amdahl M, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet. 2010;376(9752):1543-51.
de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369(26):2492-503.
de Zeeuw D, Coll B, Andress D, et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J Am Soc Nephrol. 2014;25(5):1083-93.
Wilcox G. Insulin and insulin resistance. Clin Biochem Rev. 2005;26(2):19-39.
Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest. 1994;93(2):870-6.
DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010;53(7):1270-87.
Floegel A, Stefan N, Yu Z, et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes. 2013;62(2):639-48.
Clore, JN, Allred J, White D, et al. The role of plasma fatty acid composition in endogenous glucose production in patients with type 2 diabetes mellitus. Metabolism. 2002;51(11):1471-7.
Soumura M, Kume S, Isshiki K, et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem Biophys Res Commun. 2010;402(2):265-71.
Yasuda M, Tanaka Y, Kume S, et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim Biophys Acta. 2014;1842(7):1097-108.
Koyama T, Kume S, Koya D, et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med. 2011;51(6):1258-67.
Tanaka Y, Kume S, Araki S, et al. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011;79(8):871-82.
Tanifuji C, Suzuki Y, Geot WM, et al. Reactive oxygen species-mediated signaling pathways in angiotensin II-induced MCP-1 expression of proximal tubular cells. Antioxid Redox Signal. 2005;7(9-10):1261-8.
Ruggiero C, Elks CM, Kruger C, et al. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am J Physiol Renal Physiol. 2014;306(8):F896-906.
Kamijo A, Kimura K, Sugaya T, et al. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002;62(5):1628-37.
Sasaki H, Kamijo-Ikemori A, Sugaya T, et al. Urinary fatty acids and liver-type fatty acid binding protein in diabetic nephropathy. Nephron Clin Pract. 2009;112(3):c148-56.
Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21(1):37-46.
Xia P, Inoguchi T, Kern TS, et al. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43(9):1122-9.
Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47(6):859-66.
Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135-45.
Suganami T, Yuan X, Shimoda Y, et al. Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circ Res. 2009;105(1):25-32.
Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012;15(4):518-33.
Zager RA, Johnson AC, Lund S, et al. Toll-like receptor (TLR4) shedding and depletion: acute proximal tubular cell responses to hypoxic and toxic injury. Am J Physiol Renal Physiol. 2007;292(1):F304-12.
Yang X, Zhang W, Dong M, et al. The achene mucilage hydrated in desert dew assists seed cells in maintaining DNA integrity: adaptive strategy of desert plant Artemisia sphaerocephala. PLOS ONE. 2011;6(9):e24346.
Kamijo Y, Hora K, Kono K, et al. PPARalpha protects proximal tubular cells from acute fatty acid toxicity. J Am Soc Nephrol. 2007;18(12):3089-100.
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849-61.
Davis TM, Ting R, Best JD, et al. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetologia. 2011;54(2):280-90.
Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900-17.
Takeda N, Kume S, Tanaka Y, et al. Altered unfolded protein response is implicated in the age-related exacerbation of proteinuria-induced proximal tubular cell damage. Am J Pathol. 2013;183(3):774-85.
Yoshino J, Mills KF, Yoon MJ, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528-36.
Cantó C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15(6):838-47.
Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15(6):394-409.
Tanaka Y, Kume S, Araki H, et al. 1-Methylnicotinamide ameliorates lipotoxicity-induced oxidative stress and cell death in kidney proximal tubular cells. Free Radic Biol Med. 2015;89:831-41.
Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120(4):473-82.
Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27(19):2072-85.
Madeo F, Zimmermann A, Maiuri MC, et al. Essential role for autophagy in life span extension. J Clin Invest. 2015;125(1):85-93.
Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nat Rev Endocrinol. 2009;5(7):367-73.
Wakino S, Hasegawa K, Itoh H. Sirtuin and metabolic kidney disease. Kidney Int. 2015;88(4):691-8.
Kume S, Uzu T, Horiike K, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest. 2010;120(4):1043-55.
Hasegawa K, Wakino S, Simic P, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496-504.
Kitada M, Kume S, Imaizumi N, et al. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. 2011;60(2):634-43.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728-41.
Takagi H, Matsui Y, Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal. 2007;9(9):1373-81.
Huber TB, Edelstein CL, Hartleben B, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8(7):1009-31.
Yamahara K, Kume S, Koya D, et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol. 2013;24(11):1769-81.
Cha JJ, Hyun YY, Lee MH, et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology. 2013;154(6):2144-55.
Sieber J, Lindenmeyer MT, Kampe K, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol. 2010;299(4):F821-9.
Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40(2):310-22.
Inoki K, Mori H, Wang J, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181-96.
Gödel M, Hartleben B, Herbach N, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121(6):2197-209.
Sakaguchi M, Isono M, Isshiki K, et al. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun. 2006;340(1):296-301.
Tagawa A, Yasuda M, Kume S, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. 2016;65(3):755-67.
Kume S, Koya D. Autophagy: a novel therapeutic target for diabetic nephropathy. Diabetes Metab J. 2015;39(6):451-60.
Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes. 2012;61(1):23-9.
