Corresponding author: Masafumi Matsuo, matsuo@kobe-u.ac.jp
DOI: 10.31662/jmaj.2021-0019
Received: February 13, 2021
Accepted: April 13, 2021
Advance Publication: July 9, 2021
Published: July 15, 2021
Cite this article as:
Matsuo M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021;4(3):232-240.
In 1995, we were the first to propose antisense oligonucleotide (ASO)-mediated exon-skipping therapy for the treatment of Duchenne muscular dystrophy (DMD), a noncurable, progressive muscle-wasting disease. DMD is caused by deletion mutations in one or more exons of the DMD gene that shift the translational reading frame and create a premature stop codon, thus prohibiting dystrophin production. The therapy aims to correct out-of-frame mRNAs to produce in-frame transcripts by removing an exon during splicing, with the resumption of dystrophin production. As this treatment is recognized as the most promising, many extensive studies have been performed to develop ASOs that induce the skipping of DMD exons. In 2016, an ASO designed to skip exon 51 was first approved by the Food and Drug Administration, which accelerated studies on the use of ASOs to treat other monogenic diseases. The ease of mRNA editing by ASO-mediated exon skipping has resulted in the further application of exon-skipping therapy to nonmonogenic diseases, such as diabetes mellites. Recently, this precision medicine strategy was drastically transformed for the emergent treatment of only one patient with one ASO, which represents a future aspect of ASO-mediated exon-skipping therapy for extremely rare diseases. Herein, the invention of ASO-mediated exon-skipping therapy for DMD and the current applications of ASO-mediated exon-skipping therapies are reviewed, and future perspectives on this therapeutic strategy are discussed. This overview will encourage studies on ASO-mediated exon-skipping therapy and will especially contribute to the development of treatments for noncurable diseases.
Key words: exon skipping, antisense oligonucleotide, reading frame, treatment, Duchenne muscular dystrophy
In 1995, we first proposed antisense oligonucleotide (ASO)-mediated exon-skipping therapy for Duchenne muscular dystrophy (DMD) (1), (2), and this method has since developed into a genuine strategy for DMD treatment. As exon-skipping therapy allows the editing of mRNA, it has been applied to treat both monogenic and nonmonogenic diseases. Remarkably, this precision medicine strategy was revolutionized for the treatment of a single patient, thus representing a future aspect of ASO-mediated exon-skipping therapy. In this article, ASO-mediated exon-skipping therapy is reviewed, beginning from its invention for DMD treatment to its wider uses in the treatments of nonmonogenic diseases, and future perspectives for ASO-mediated exon-skipping therapies are discussed.
Exon skipping involves the removal of certain exons from mRNAs during splicing, during which introns are removed from pre-mRNA to produce mature mRNA. Although splicing is strictly regulated to produce mRNAs with no errors, alternative splicing occurs in more than 90% of human genes to produce a variety of mRNAs and diverse protein products (3). Notably, exon skipping is the most common pattern of alternative splicing. In cancer cells, for example, 90,616 exon-skipping events are reported to occur in 14,272 genes (4).
On the other hand, exon skipping is induced by gene mutations and causes genetic diseases. Exons are defined by splicing consensus sequences located at the junction of exons and introns in pre-mRNA, and GT and AG dinucleotides are strictly conserved at the splice donor and acceptor sites, respectively. Furthermore, this definition is supported by the splicing cis elements located in exon or intron sequences, and most mutations that induce exon skipping are located at splicing consensus sequences. In addition, mutations splicing regulatory cis elements, such as exonic splicing enhancers/silencers and intronic splicing enhancers/silencers, cause exon skipping, and exon-skipping mutations are common in genetic diseases (5).
DMD (OMIM 310200), a fatal X chromosome-linked recessive disorder, is the most common inherited muscle disease and affects one in 5,000 to 6,000 live-born male children. DMD is characterized by infantile-onset progressive muscle wasting, and patients usually succumb to the disease during their 20s. Although DMD has remained uncurable since its initial appearance in 1868, the life expectancy of patients with DMD has been extended slowly by general medical treatments and physical therapy (6), (7), and establishing a fundamental treatment protocol has long been the goal of DMD studies.
In 1987, the DMD gene, responsible for DMD, was cloned (8) and shown to comprise 79 exons spanning 2.4 Mb on the X chromosome and to yield a 14-kb cDNA encoding dystrophin; dystrophin deficiency leads to DMD. Exon deletions, which are the most common deletion type (>50%), cause both DMD and Becker muscular dystrophy (BMD), a milder progressive muscle-wasting disease. The differences in the two types of diseases are attributed to a translational reading frame rule (9); mutations shifting the translational reading frame of the mRNA usually result in the more severe DMD phenotype because of the creation of premature stop codons that totally prohibit dystrophin protein expression, whereas mutations that maintain the original mRNA reading frame cause the milder BMD phenotype because a mutated but still functional dystrophin protein can be expressed from the mRNA.
A case of a Japanese boy with DMD contributed greatly to the development of ASO-mediated exon-skipping therapy (10). For this patient, a small product of exon 19 of the DMD gene was produced by polymerase chain reaction (PCR), and the sequencing of the product revealed a 52-bp deletion within the 88-bp exon 19, thus shortening the exon to 36 bp (11). This out-of-frame genomic deletion in DMD is 6 bp upstream of the 3′ end of exon 19 and does not disrupt the splicing consensus sequences, consistent with the reading frame rule (9). This mutation caused the smallest deletion known at the time and was termed dystrophin Kobe (11).
Analysis of the patient’s DMD cDNA revealed a complete absence of the exon 19 sequence, indicating the skipping of exon 19, although the consensus sequences at the splicing acceptor and donor sites were intact. Remarkably, the cause of exon skipping was attributed to the deletion of an exonic splicing enhancer encoded by the deleted sequence (1). This was the first demonstration of an exonic splicing enhancer being critical for the recognition of DMD gene exons. Furthermore, ASOs directed against this splicing enhancer sequence were shown to inhibit the splicing of exon 19 (1).
Based on the above findings, we proposed a novel precision medicine strategy for the use of ASO-mediated exon-skipping therapy for DMD. The induction of exon skipping allows one exon to be omitted from the mutated DMD mRNA, leading to the conversion of an out-of-frame mRNA to an in-frame mRNA, thereby restoring dystrophin production (1), (2). Subsequently, we showed that an ASO against exon 19 could induce the skipping of exon 19 of the endogenous transcript (12).
Furthermore, we obtained convincing evidence for the efficacy of exon-skipping therapy on a Japanese patient with BMD exhibiting a single-nucleotide mutation in exon 27 (c.3631G > T) that led to the substitution of glutamate for a stop codon (E1211X) (13). The single-nucleotide mutation induced the skipping of exon 27, thereby producing an in-frame semifunctional DMD mRNA sequence and leading to the production of a truncated dystrophin protein, with a mild disease phenotype. These results clearly indicate the potential effect of exon skipping as a treatment for DMD.
Theoretically, ASOs capable of inducing exon 19 skipping should produce in-frame mRNAs in patients with DMD carrying an exon 20 deletion. This was verified by the transfection of an ASO into cultured muscle cells obtained from a patient with DMD with a deletion of exon 20 (14). Subsequently, the patient received the ASO via intravenous infusion once a week for 4 weeks (15), and a muscle biopsy performed at 1 week after the final infusion revealed a novel in-frame mRNA lacking both exon 19 and exon 20 that represented approximately 6% of the total reverse transcription PCR product. Dystrophin was identified histochemically in the sarcolemma of muscle cells after ASO treatment. These findings demonstrate that ASO-mediated exon skipping shows promise for the treatment of DMD.
The DMD gene contains numerous hotspots for exon deletion. Among exon deletions in patients with DMD, approximately 20%, 13%, 12%, and 11% are amendable with the skipping of exons 51, 53, 45, and 44, respectively (16). To treat a sufficient number of patients with DMD, several ASOs targeting the top three exons have been developed for clinical use by international researchers. As a result, eteplirsen, a 25-mer phosphorodiamidate morpholino ASO injected intravenously, was the first approved by the Food and Drug Administration (FDA) in 2016 for the treatment of patients with DMD with mutations amenable to the skipping of exon 51 (Table 1). Golodirsen, a morpholino ASO targeting the skipping of exon 53, was next approved in 2019 (17), and viltolarsen, a 21-mer phosphorodiamidate morpholino ASO targeting the skipping of exon 51, was approved in 2020 (18).
Table 1. Antisense Oligonucleotides (ASOs) to Treat Duchenne Muscular Dystrophy (DMD).
| Name | ASO | Target exon | Phase | ||
|---|---|---|---|---|---|
| Nucleic acid | Size | Route | |||
| Eteplirsen | PMO | 25 | IV | 51 | Approved |
| Golodirsen | PMO | 25 | IV | 53 | Approved |
| Viltolarsen | PMO | 21 | IV | 53 | Approved |
| Casimersen | PMO | 25 | IV | 45 | Approval stage |
| Renadirsen | ENA/2'RNA | 18 | SC | 45 | I/II |
| PMO: phosphorodiamidate morpholino; ENA: 2'-O,4'-C-ethylene-bridged nucleic acid; 2'RNA: 2'-O-methyl RNA; IV: intravenous injection; SC: subcutaneous injection | |||||
We developed an 18-mer ASO that induces the skipping of exon 45, named renadirsen, that contains both 2'-O,4'-C-ethylene-bridged nucleic acid (ENA) and 2'-O-methyl RNA (19). A modified ENA invented in Japan was shown to have suitable characteristics for clinical use, such as high affinity for complementary sequences and high resistance to nuclease (20). Renadirsen was injected subcutaneously into patients in a phase I/II study (JapicCTI-153072).
ASO-mediated exon skipping serves as a method to edit mRNA and can therefore be applied to manipulate gene functions for restoration, destruction, and modulation (Table 2). For the restoration of gene function, mRNAs that lose certain exons should be functional or semifunctional, and this strategy is applied mainly to induce the skipping of mutated exons in monogenic diseases. In contrast, ASO-mediated exon skipping is applied for the disruption of gene function, which involves the targeting of out-of-frame exons, and this strategy has been applied to suppress MSTN gene function, thereby promoting muscle growth. In particular, the skipping of in-frame exons has been implemented to modulate gene function, producing an isoform that differs in structure and function from the original product. This strategy has also been applied to the removal of an integrin alpha 4 transmembrane domain by the skipping of exon 27 of the ITG4 gene for the treatment of Crohn’s disease. Although exon skipping has multiple potential applications, caution should be taken to avoid the unintentional skipping of flanking exons, which has been reported in the ITGA4 and COL7A1 genes (21), (22).
Table 2. Gene Functional Changes Induced by Exon Skipping.
| Function | Target exon | Gene | Inheritance | Disease |
|---|---|---|---|---|
| Restoration | Normal out-of-frame | DMD | Monogenetic | DMD |
| Mutated in-frame exon | DYSF | Monogenetic | LGMD2B | |
| Mutated out-of-frame exon | SGCG | Monogenetic | LGMD2C | |
| Mutated pseudo exon | CLRN1 | Monogenetic | USH3 | |
| Destruction | Out-of-frame exon | MSTN | Non monogenetic | Diabetes mellitus |
| Modulation | Normal in-frame exon | ITGA4 | Non monogenetic | Crohn’s disease |
| DMD: Duchenne muscular dystrophy; LGMD2B: limb girdle muscular dystrophy 2B; LGMD2C: limb girdle muscular dystrophy 2C; USH3: Usher syndrome type III | ||||
The skipping of out-of-frame exons shifts the translational reading frame and is therefore applied to the conversion of out-of-frame mutations to in-frame mutations, as shown in DMD therapy (Figure 1 and Table 3). For dysferlinopathy and Parkinson’s disease, ASOs were designed for the skipping of DYSF and PRKN exons to restore the reading frame for the production of semifunctional proteins (23), (24).
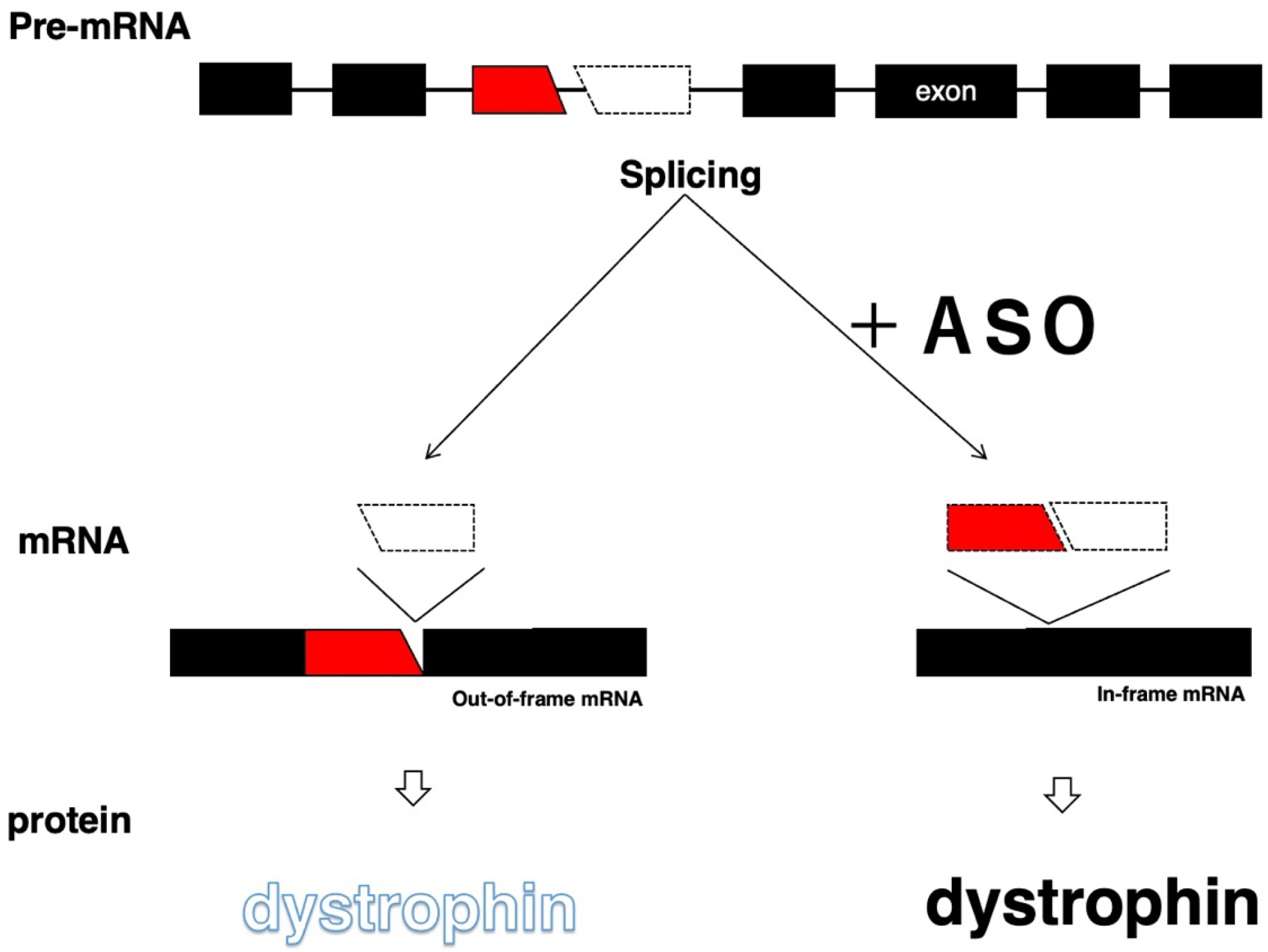
Table 3. Skipping of Normal Out-of-frame Exons.
The skipping of an in-frame exon encoding a disease-causing mutation aims to remove the mutation and produce in-frame mRNA, which should give rise to the semifunctional truncated protein. Therefore, this method can be applied to cases in which the truncated protein is confirmed to be semifunctional (Table 4). ASOs targeting mutated exons of the DYSF and LMNA genes have been studied to restore dysferlin in limb girdle muscular dystrophy 2B and laminin in laminopathy, respectively (26), (27). As most collagen molecules are synthesized in a trimeric procollagen form, exon-skipping therapies producing truncated proteins is challenging for the treatment of collagenopathy (28). Despite this, an ASO targeting exon 21 encoding a nonsense mutation of the COL4A5 gene successfully improved the clinical phenotypes of a mouse model of Alport syndrome (29), suggesting that mutations in the in-frame exons of collagen genes are amendable by exon skipping. At the same time, an ASO designed to induce the skipping of exon 73 of the COL7A1 gene, a mutational hotspot, was developed for the treatment of dystrophic epidermolysis bullosa (30). For dystrophic epidermolysis bullosa, ASOs can be administered topically, opening a new avenue for ASO transfection (30). ASO-mediated exon-skipping therapies have also been applied to several other diseases, including enzyme deficiency (Table 4).
Table 4. Skipping of Mutated In-frame Exons.
| Disease | Gene | Exon | Phase | Author, Year |
|---|---|---|---|---|
| LGMD2B | DYSF | 32 | P | Barthelemy et al. 2015 (26) |
| Laminopathy | LMNA | 5 | P | Scharner et al. 2015 (27) |
| Joubert syndrome | CEP290 | 41 | P | Molinari et al. 2019 (31) |
| Alport syndrome | COL4A5 | 21 | P | Yamamura et al. 2020 (29) |
| Dystrophic epidermolysis bullosa | COL7A1 | 73 | P | Bornert et al. 2020 (30) |
| CADASIL | NOTCH3 | 9 | P | Gravesteijn et al. 2020 (32) |
| Mucolipidosis II | GNPTAB | 19 | P | Matos et al. 2020 (33) |
| Myotonic dystrophy type1 | DMPK | 15 | P | Stepniak et al. 2020 (34) |
| LGMD2B: limb girdle muscular dystrophy 2B; CADASIL: cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; P: preclinical study | ||||
The skipping of mutated out-of-frame exons can remove the mutations but produce out-of-frame mRNAs incapable of producing proteins. Therefore, this strategy is applied in three situations: (1) the skipping of multiple exons to produce in-frame mRNA; (2) the skipping of a single exon to reduce a certain isoform; and (3) the skipping of the penultimate exon (Table 5). For example, four exons of the SGCG gene, including an out-of-frame exon with a mutation, were removed together using a combination of ASOs for the treatment of limb girdle muscular dystrophy type 2C (35). The skipping of an exon to reduce a certain isoform was applied in the treatment of laminopathy, which reduced laminin A production and thus led to the reduced accumulation of a toxic substance (36). The skipping of the penultimate exon has been investigated for the treatment of spinocerebellar ataxia type 3 by skipping exon 10 of the ATXN3 gene (37).
Table 5. Skipping of Mutated Out-of-frame Exons.
| Disease | Gene | Exon | Phase | Author, Year |
|---|---|---|---|---|
| LGMD2C | SGCG | 4-7 | P | Wyatt et al. 2018 (35) |
| CADASIL | NOTCH3 | 2-3,4-5,6 | P | Rutten 2016 (38) |
| HGPS | LMNA | 11 | P | Lee et al. 2016 (36) |
| SCA3 | ATXN3 | 10 | P | McIntosh et al. 2019 (37) |
| LGMD2C: limb girdle muscular dystrophy 2C; CADASIL: cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; HPGS: Hutchinson-Gilford progeria syndrome; SCA3: spinocerebellar ataxia type 3; P: preclinical study | ||||
The skipping of mutated pseudoexons is expected to produce completely normal mRNAs and proteins and is therefore considered a complete treatment. One study revealed that pseudoexons were incorporated by the creation of 5' and 3' splice sites in 52 and 11 genes, respectively, and by the interference of splicing regulatory elements in nine genes (39). In cancer, pseudoexons have been identified in 10 genes directly involved in cancer pathology, such as ATM, BRCA1, and NF1, and in 32 cancer-related genes, such as CYBB and CYP17A1 (40). Every pseudoexon can be a target of skipping therapy, and recent studies on pseudoexon skipping are summarized in Table 6. Nevertheless, mutations that create identical pseudoexons are rarely reported, and the low patient numbers have hampered the clinical development of ASOs designed for this purpose.
Table 6. Skipping of Pseudoexons.
| Disease | Gene | Phase | Author, Year |
|---|---|---|---|
| Usher syndrome type III | CLRN1 | P | Panagiotopoulos et al. 2020 (41) |
| LGMD2B | DYSF | P | Dominov et al. 2020 (42) |
| Erythropoietic protoporphyria | FECH | P | Halloy et al. 2020 (43) |
| Stargardt disease | ABCA4 | P | Sangermano et al. 2019 (44) |
| Neuronal ceroid lipofuscinosis 7 | CLN7 | H | Kim et al. 2019 (45) |
| LGMD2B: limb girdle muscular dystrophy 2B; P: preclinical study; H: human trial | |||
The skipping of out-of-frame exons is a gene silencing method that involves the alteration of the reading frame and has therefore been applied to treat diseases involving modifier genes (Table 7). As gene function can be silenced via techniques such as ASO-mediated RNase and siRNA transfection (46), ASO-mediated exon skipping has not been well studied for the treatment of diseases. An ASO designed to induce the skipping of the out-of-frame exon 2 of the MSTN gene was studied to enhance muscle growth in patients with muscular dystrophies and diabetes mellitus (47). An ASO designed to induce the skipping of exon 41 of LRRK2, which encodes part of the kinase domain, was developed to reduce LRRK2 kinase function in patients with genetic and sporadic Parkinson’s disease (48). Interestingly, an ASO designed to induce the skipping of the out-of-frame exon 4 of the fusion gene ERG was developed to suppress its function in prostate cancer cells and markedly reduced ERG protein levels, resulting in decreased cell proliferation (49).
Table 7. Skipping of Out-of-frame Exons for the Disruption of Gene Function.
| Disease | Gene | Exon | Phase | Author, Year |
|---|---|---|---|---|
| Diabetes mellitus | MSTN | 2 | P | Eilers et al. 2021 (50) |
| DMD | P | Kemaladewi et al. 2011 (47) | ||
| Parkinson’s disease | LRRK2 | 41 | P | Korecka et al. 2020 (48) |
| Prostate cancer | ERG | 4 | P | Li et al. 2020 (49) |
| Muscle fibrosis | ALK5 | 6 | P | Kemaladewi et al. 2014 (51) |
| Familial hypercholesterolemia | APOB | 27 | P | Disterer et al. 2013 (52) |
| DMD: Duchenne muscular dystrophy; P: preclinical study | ||||
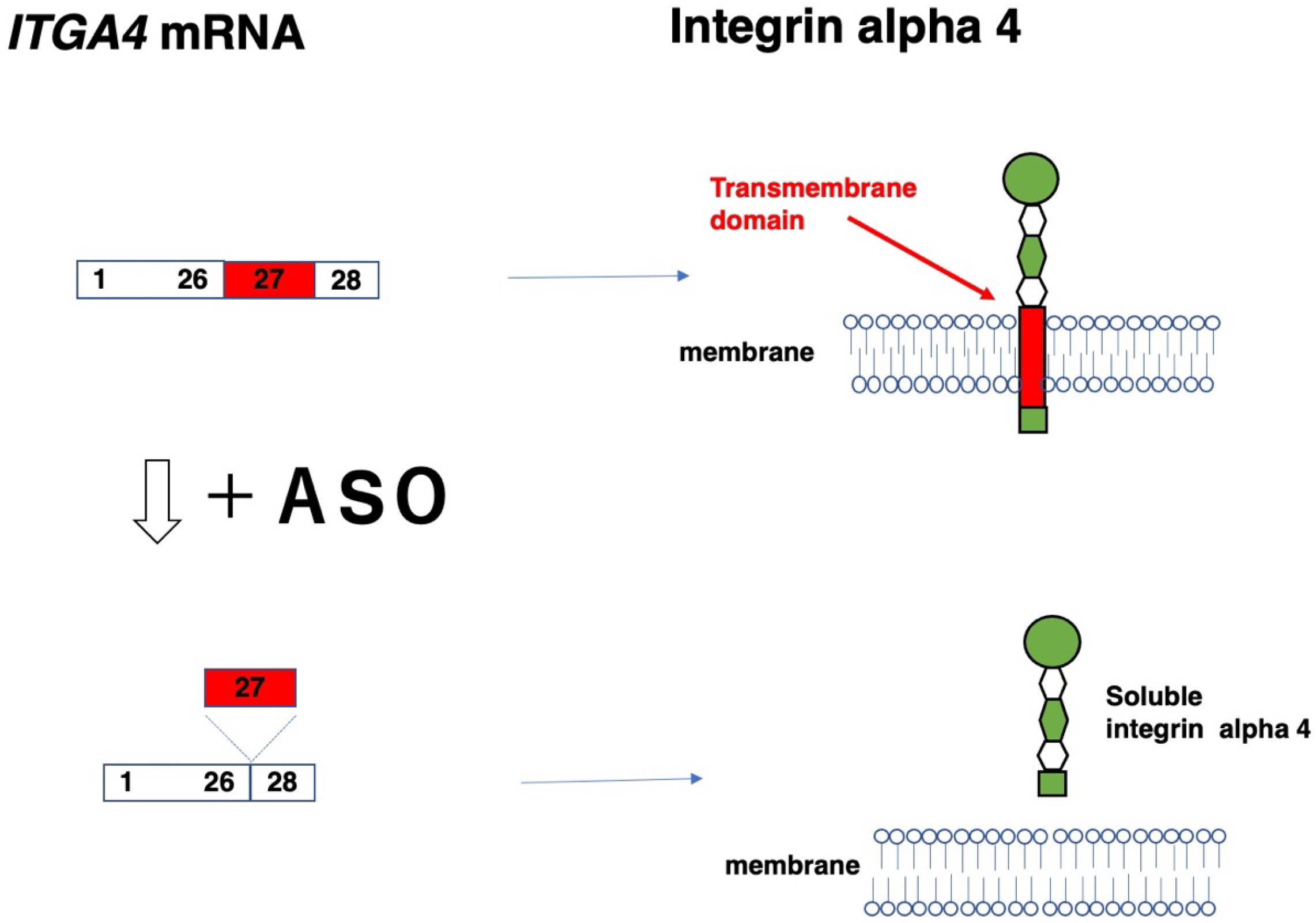
The skipping of in-frame exons produces in-frame mRNAs, thereby resulting in the production of short, truncated proteins. In cases of truncated proteins functioning differently from the original protein products, this exon skipping is applied to modulate gene function (Table 8). One example was reported in the ITGA4 gene (21). Moreover, exon 27 was removed from the normal gene to produce an integrin alpha 4 protein lacking a transmembrane domain (Figure 2), resulting in the production of soluble integrin alpha 4 that bound beta. This strategy was devised to treat diseases such as Crohn’s disease and multiple sclerosis, which are modified by the hyperactivity of integrin beta.
Table 8. Skipping of Normal In-frame Exons.
| Disease | Gene | Exon | Lost domain | Phase | Author, Year |
|---|---|---|---|---|---|
| Crohn’s disease | ITGA4 | 27 | transmembrane | P | Aung-Htut 2019 (21) |
| Dupuytren’s disease | ALK5 | 2 | ligand binding | P | Karkampouna et al. 2014 (53) |
| Muscle fibrosis | P | Kemaladewi et al. 2014 (51) | |||
| Hypertrophic scar | P | Raktoe et al. 2020 (54) | |||
| Cancer | CD44 | v8 | ligand binding | P | Fukushima et al. 2020 (55) |
| P: preclinical study |
ASO-mediated exon-skipping therapies are now applicable for a wide variety of diseases. Currently, most exon-skippable ASOs under clinical development have been developed to treat a certain number of patients, and ASOs designed to induce the skipping of exons 51, 53, and 45 have been developed for the clinical treatment of patients with DMD. In contrast, patients with deletions not treatable with these ASOs do not benefit from these treatment opportunities, even though exon skipping is theoretically applicable. Recently, one ASO was developed for the treatment of one patient within a remarkably short time period to stop the aggressive progression of a neurodegenerative disease (Table 6) (45). This event stimulated a large discussion on precision medicine among staff at the FDA (56), (57), and the one-drug/one-patient policy may enhance the further development of ASOs and open a new avenue for the treatment of patients, especially those with rare uncurable diseases.
ASO-mediated exon-skipping therapy is an emerging area of drug development that targets the disease source at the RNA level and offers a promising therapy for a wide variety of diseases. However, there are some concerns about the therapy: first, side effects including off-target effects should be monitored carefully, because ultra-long-term treatment has not yet been completed. Second, currently approved ASOs are expensive. These high costs are not specific to ASOs but relate to the fact that ASOs mainly target rare diseases. As the number of treated patients is lower, development costs per patient are higher (58). The cost to synthesize ASO would be reduced by a large-scale synthesis when the demand for ASOs increases by treating common diseases. Third, the delivery of ASOs has always been a significant hurdle for their broad clinical applications, and various strategies have been employed to deliver them (59). For example, the conjugation of ASO to N-acetyl galactosamine (GalNAc) is a well-defined liver-targeted moiety, because GalNAc selectively targets the asialoglycoprotein receptor that is highly expressed in hepatocytes (60). These concerns will be solved by conducting more studies on ASO drugs in the future.
Masafumi Matsuo discloses being employed by Kobe Gakuin University, which received funding from KNC Laboratories Inc. (Kobe, Japan). The author further discloses being a scientific adviser for Daiichi-Sankyo Co. (Tokyo, Japan) and JCR Pharma Co. (Ashiya, Japan).
This work was partly supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED grant number [20ek0109442h 0001].
Takeshima Y, Nishio H, Sakamoto H, et al. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J Clin Invest. 1995;95(2):515-20.
Matsuo M. Duchenne/Becker muscular dystrophy: from molecular diagnosis to gene therapy. Brain Dev. 1996;18(3):167-72.
Pan Q, Shai O, Lee LJ, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413-5.
Kim P, Yang M, Yiya K, et al. ExonSkipDB: functional annotation of exon skipping event in human. Nucleic Acids Res. 2020;48(D1):D896-907.
Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19-32.
Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12(10):926-9.
Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012;31(2):121-5.
Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509-17.
Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90-5.
Matsuo M, Takeshima Y, Nishio H. Contributions of Japanese patients to development of antisense therapy for DMD. Brain Dev. 2016;38(1):4-9.
Matsuo M, Masumura T, Nakajima T, et al. A very small frame-shifting deletion within exon 19 of the Duchenne muscular dystrophy gene. Biochem Biophys Res Commun. 1990;170(2):963-7.
Pramono ZA, Takeshima Y, Alimsardjono H, et al. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem Biophys Res Commun. 1996;226(2):445-9.
Shiga N, Takeshima Y, Sakamoto H, et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest. 1997;100(9):2204-10.
Takeshima Y, Wada H, Yagi M, et al. Oligonucleotides against a splicing enhancer sequence led to dystrophin production in muscle cells from a Duchenne muscular dystrophy patient. Brain Dev. 2001;23(8):788-90.
Takeshima Y, Yagi M, Wada H, et al. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr Res. 2006;59(5):690-4.
Flotats BM, Hahn A. New therapeutics options for pediatric neuromuscular disorders. Front Pediatr. 2020;8:583877.
Heo YA. Golodirsen: first approval. Drugs. 2020;80(3):329-33.
Komaki H, Takeshima Y, Matsumura T, et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: a phase 1/2 study. Ann ClinTransl Neurol. 2020;7(12):2393-408.
Lee T, Awano H, Yagi M, et al. 2'-O-methyl RNA/ethylene-bridged nucleic acid chimera antisense oligonucleotides to induce dystrophin exon 45 skipping. Genes. 2017;8(2):67.
Koizumi M. 2'-O,4'-C-ethylene-bridged nucleic acids (ENA) as next-generation antisense and antigene agents. Biol Pharm Bull. 2004;27(4):453-6.
Aung HM, Comerford I, Johnsen R, et al. Reduction of integrin alpha 4 activity through splice modulating antisense oligonucleotides. Sci Rep. 2019;9:12994.
Ham KA, Aung-Htut MT, Fletcher S, et al. Nonsequential splicing events alter antisense-mediated exon skipping outcome in COL7A1. Int J Mol Sci. 2020;21(20):7705.
Verwey N, Gazzoli I, Krause S, et al. Antisense-mediated skipping of dysferlin exons in control and dysferlinopathy patient-derived cells. Nucleic Acid Ther. 2020;30(21):71-9.
Li D, Aung-Htut MT, Ham KA, et al. A Splice intervention therapy for autosomal recessive juvenile Parkinson's disease arising from parkin mutations. Int J Mol Sci. 2020;21(19):7282.
Schneider AF, Aartsma RA. Developments in reading frame restoring therapy approaches for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2021;21(3):343-59.
Barthélémy F, Blouin C, Wein N, et al. Exon 32 skipping of dysferlin rescues membrane repair in patients' cells. J Neuromuscul Dis. 2015;2(3):281-90.
Scharner J, Figeac N, Ellis JA, et al. Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon-skipping therapy for laminopathies. Gene Ther. 2015;22(6):503-15.
Li D, Mastaglia F, Fletcher S, et al. Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol Sci. 2018;39(11):982-94.
Yamamura T, Horinouchi T, Adachi T, et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat Commun. 2020;11(1):2777.
Bornert O, Hogervorst M, Nauroy P, et al. QR-313, an antisense oligonucleotide, shows therapeutic efficacy for treatment of dominant and recessive dystrophic epidermolysis bullosa: a preclinical study. J Invest Dermatol. 2021;141(4):883-93.e6.
Molinari E, Ramsbottom S, Srivastava S, et al. Targeted exon skipping rescues ciliary protein composition defects in Joubert syndrome patient fibroblasts. Sci Rep. 2019;9(1):10828.
Gravesteijn G, Dauwerse J, Overzier M, et al. Naturally occurring NOTCH3 exon skipping attenuates NOTCH3 protein aggregation and disease severity in CADASIL patients. Hum Mol Genet. 2020;29(11):1853-63.
Matos L, Vilela R, Rocha M, et al. Development of an antisense oligonucleotide-mediated exon skipping therapeutic strategy for mucolipidosis II: validation at RNA level. Hum Gene Ther. 2020;31(13-14):775-83.
Stepniak-Konieczna E, Konieczny P, Cywoniuk P, et al. AON-induced splice-switching and DMPK pre-mRNA degradation as potential therapeutic approaches for Myotonic Dystrophy type 1. Nucleic Acids Res. 2020;48(5):2531-43.
Wyatt EJ, Demonbreun AR, Kim EY, et al. Efficient exon skipping of SGCG mutations mediated by phosphorodiamidate morpholino oligomers. JCI Insight. 2018;3(9):e99357.
Lee JM, Nobumori C, Tu Y, et al. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016;126(4):1592-602.
McIntosh CS, Aung-Htut MT, Fletcher S, et al. Removal of the polyglutamine repeat of ataxin-3 by redirecting pre-mRNA processing. Int J Mol Sci. 2019;20(21):5434.
Rutten JW, Dauwerse HG, Peters DJM, et al. Therapeutic NOTCH3 cysteine correction in CADASIL using exon skipping: in vitro proof of concept. Brain. 2016;139(4):1123-35.
Vaz-Drago R, Custódio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet. 2017;136(9):1093-111.
Romano M, Buratti E, Baralle D. Role of pseudoexons and pseudointrons in human cancer. Int J Cell Biol. 2013;2013:810572.
Panagiotopoulos AL, Karguth N, Pavlou M, et al. Antisense oligonucleotide- and CRISPR-Cas9-mediated rescue of mRNA splicing for a deep intronic CLRN1 mutation. Mol Ther Nucleic Acids. 2020;21:1050-61.
Dominov J, Uyan Ö, McKenna-Yasek D, et al. Correction of pseudoexon splicing caused by a novel intronic dysferlin mutation. Ann Clin Transl Neurol. 2019;6:642-54.
Halloy F, Iyer P, Ćwiek P, et al. Delivery of oligonucleotides to bone marrow to modulate ferrochelatase splicing in a mouse model of erythropoietic protoporphyria. Nucleic Acids Res. 2020;48(9):4658-71.
Sangermano R, Garanto A, Runhart E, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21(8):1751-60.
Kim J, Hu C, Moufawad El Achkar C, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381(17):1644-52.
Fuchs U, Damm-Welk C, Borkhardt A. Silencing of disease-related genes by small interfering RNAs. Curr Mol Med. 2004;4(5):507-17.
Kemaladewi DU, Hoogaars WM, van Heiningen SH, et al. Dual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophy. BMC Med Genomics. 2011;4(1):36.
Korecka JA, Thomas R, Hinrich AJ, et al. Splice-switching antisense oligonucleotides reduce LRRK2 kinase activity in human LRRK2 transgenic mice. Mol Ther Nucleic Acids. 2020;21:623-35.
Li L, Hobson L, Perry L, et al. Targeting the ERG oncogene with splice-switching oligonucleotides as a novel therapeutic strategy in prostate cancer. Brit J Cancer. 2020;123(6):1024-32.
Eilers W, Cleasby M, Foster K. Development of antisense-mediated myostatin knockdown for the treatment of insulin resistance. Sci Rep. 2021;11(1):1604.
Kemaladewi D, Pasteuning S, van der Meulen JW, et al. Targeting TGF-β signaling by antisense oligonucleotide-mediated knockdown of TGF-β type I receptor. Mol Ther Nucleic Acids. 2014;3:e156.
Disterer P, Al Shawi R, Ellmerich S, et al. Exon skipping of hepatic APOB pre-mRNA with splice-switching oligonucleotides reduces LDL cholesterol in vivo. Mol Ther. 2013;21(3):602-9.
Karkampouna S, Kruithof B, Kloen P, et al. Novel ex vivo culture method for the study of Dupuytren's disease: effects of TGFβ type 1 receptor modulation by antisense oligonucleotides. Mol Ther Nucleic Acids. 2014;3:e142.
Raktoe RS, Rietveld MH, Out-Luiting J, et al. Exon skipping of TGFβRI affects signalling and ECM expression in hypertrophic scar-derived fibroblasts. Scars Burn Heal. 2020;6:2059513120908857.
Fukushima S, Farea M, Maeta K, et al. Dual fluorescence splicing reporter minigene identifies an antisense oligonucleotide to skip exon v8 of the CD44 gene. Int J Mol Sci. 2020;21(23):E9316.
Fyfe I. Oligonucleotide designed for ultimate personalized treatment. Nat Rev Neurol. 2019;15(12):687.
Woodcock J, Marks P. Drug regulation in the era of individualized therapies. N Engl J Med. 2019;381(17):1678-80.
Kuijper E, Bergsma A, Pijnappel WWMP, et al. Opportunities and challenges for antisense oligonucleotide therapies. J Inherit Metab Dis. 2021;44(1):72-87.
Dhuri K, Bechtold C, Quijano E, et al. Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. 2020;9(6):2004.
Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol Ther Nucleic Acids. 2017;6:116-32.
