Corresponding author: Noboru Mizushima, nmizu@m.u-tokyo.ac.jp
DOI: 10.31662/jmaj.2021-0090
Received: May 10, 2021
Accepted: May 13, 2021
Advance Publication: July 6, 2021
Published: July 15, 2021
Cite this article as:
Yamamoto Y, Mizushima N. Autophagy and Ciliogenesis. JMA J. 2021;4(3):207-215.
Autophagy is a major intracellular degradation system and plays important roles in various physiological processes such as metabolic adaptation and intracellular homeostasis. It degrades intracellular components both randomly and selectively. Autophagic activity is tightly regulated primarily by nutrient availability, but also by other extracellular and intracellular signals. Growing evidence suggests that there are multiple links between autophagy and the primary cilium. The primary cilium is an organelle present on the cell surface and is important for keeping cellular integrity by transducing extracellular stimuli inside the cell. Recent studies have revealed that autophagy selectively degrades the ciliogenesis inhibitory proteins OFD1 and MYH9, promoting ciliogenesis. Conversely, autophagy also inhibits ciliogenesis under growth conditions. The primary cilium can also regulate autophagic activity. These findings suggest that the relationship between autophagy and the primary cilia is bidirectional, and that both are important for maintaining the normal function of various organs.
Key words: autophagy, ciliogenesis, ciliopathy
Macroautophagy (hereinafter, autophagy) is an intracellular degradation process by which cytoplasmic components are degraded in the lysosome (Figure 1a). Autophagy plays key roles in various physiological processes including metabolic adaptation, development, and tissue homeostasis as well as in pathological conditions such as neurodegenerative diseases, muscle and liver diseases, and cancers (1), (2). When autophagy is induced, contents of the cytoplasm are enclosed by a small membrane sac called the isolation membrane. When the membrane is closed, it becomes a double membrane structure called the autophagosome (Figure 1a) (3), (4). The autophagosome then fuses with lysosomes to degrade the engulfed material. Autophagy is typically induced by nutrient starvation, which is mainly mediated by the mechanistic target of rapamycin (mTOR), a serine-threonine kinase that positively regulates cell growth and proliferation by promoting anabolic activities, and negatively regulates autophagy by inhibiting upstream autophagy regulators (5).
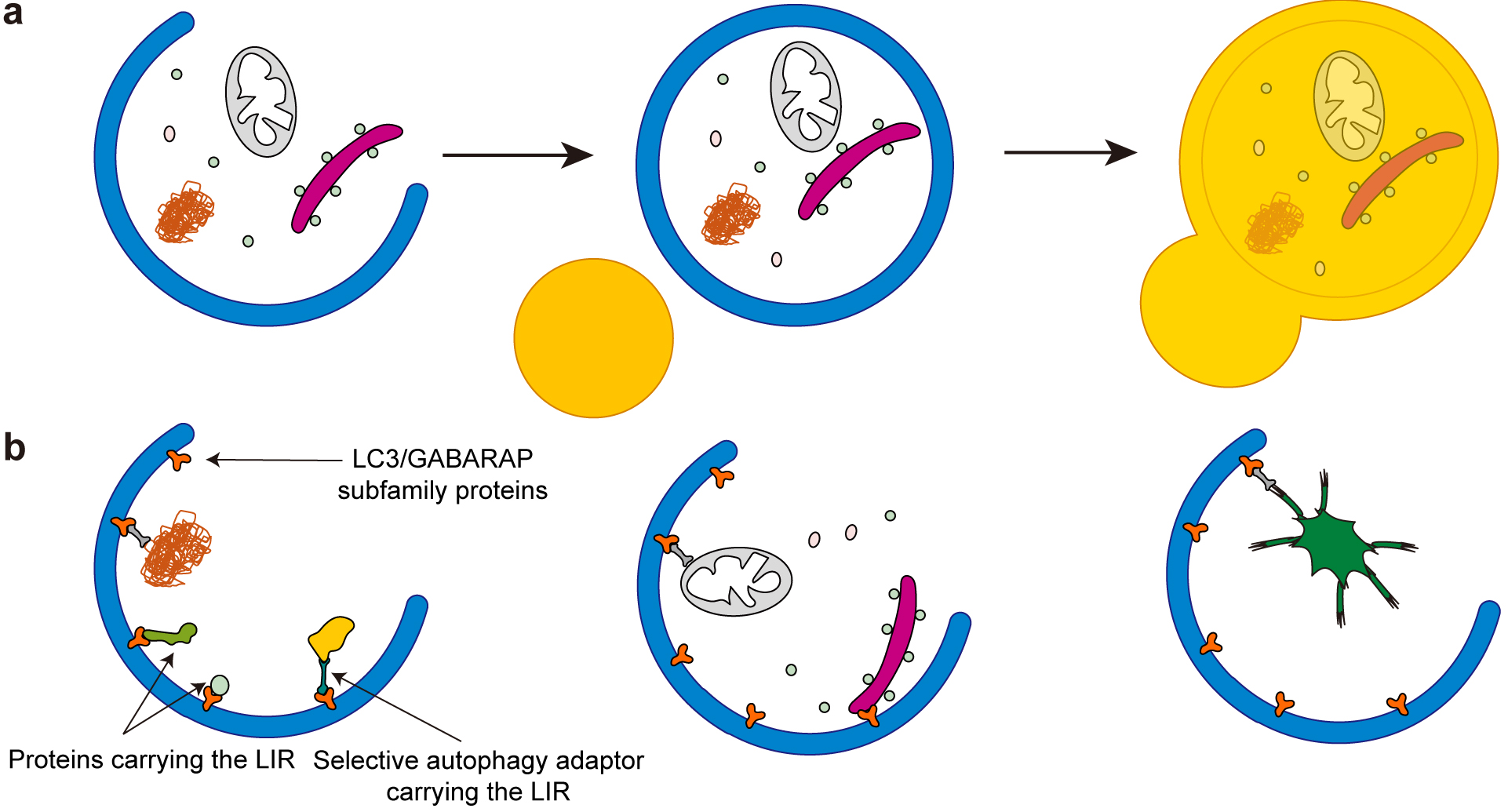
Although autophagy was previously viewed as primarily a non-selective intracellular degradation system, recent studies have shown that autophagy also degrades certain substrates selectively (Figure 1b) (6), (7). Cargo (or substrates) of selective autophagy are soluble and aggregated proteins; organelles (often damaged), including mitochondria, the endoplasmic reticulum, peroxisomes, and lysosomes; and even intracellular pathogens (8), (9), (10), (11), (12). Most of this cargo is recognized by autophagosomal proteins via ATG8 family proteins, which are classified into the following two subfamilies in mammals: LC3 (including LC3A, LC3B, and LC3C) and GABARAP (including GABARAP, GABARAPL1, and GABARAPL2). These ATG8 family proteins are covalently conjugated to phosphatidylethanolamine in the autophagic membrane and bind to selective cargo that has a motif called the LC3-interacting region (LIR) (Figure 1b) (13), (14). In some cases, cargo is indirectly recognized by ATG8 proteins through LIR-containing selective autophagy adaptors such as SQSTM1 (also called p62), NBR1, NDP52, optineurin, and TAX1BP1, which interact with both ATG8 and selective autophagy cargo simultaneously (6), (7). These selective autophagy adaptors are also degraded along with their cargo by autophagy.
The primary cilium is a hair-like, microtubule-based organelle present on the surface of almost all vertebrate cells (15). It is composed of a basal body, a matured form of the mother centriole of the centrosome, and a microtubule-based structure called the axoneme (Figure 2). At the base of the cilium, there is a transition zone that serves as a barrier to tightly regulate protein entry to and exit from the ciliary compartments. Inside the cilium, ciliary proteins are transported back and forth by intraflagellar transport (IFT) machinery, including various kinesins and dyneins (Figure 2).
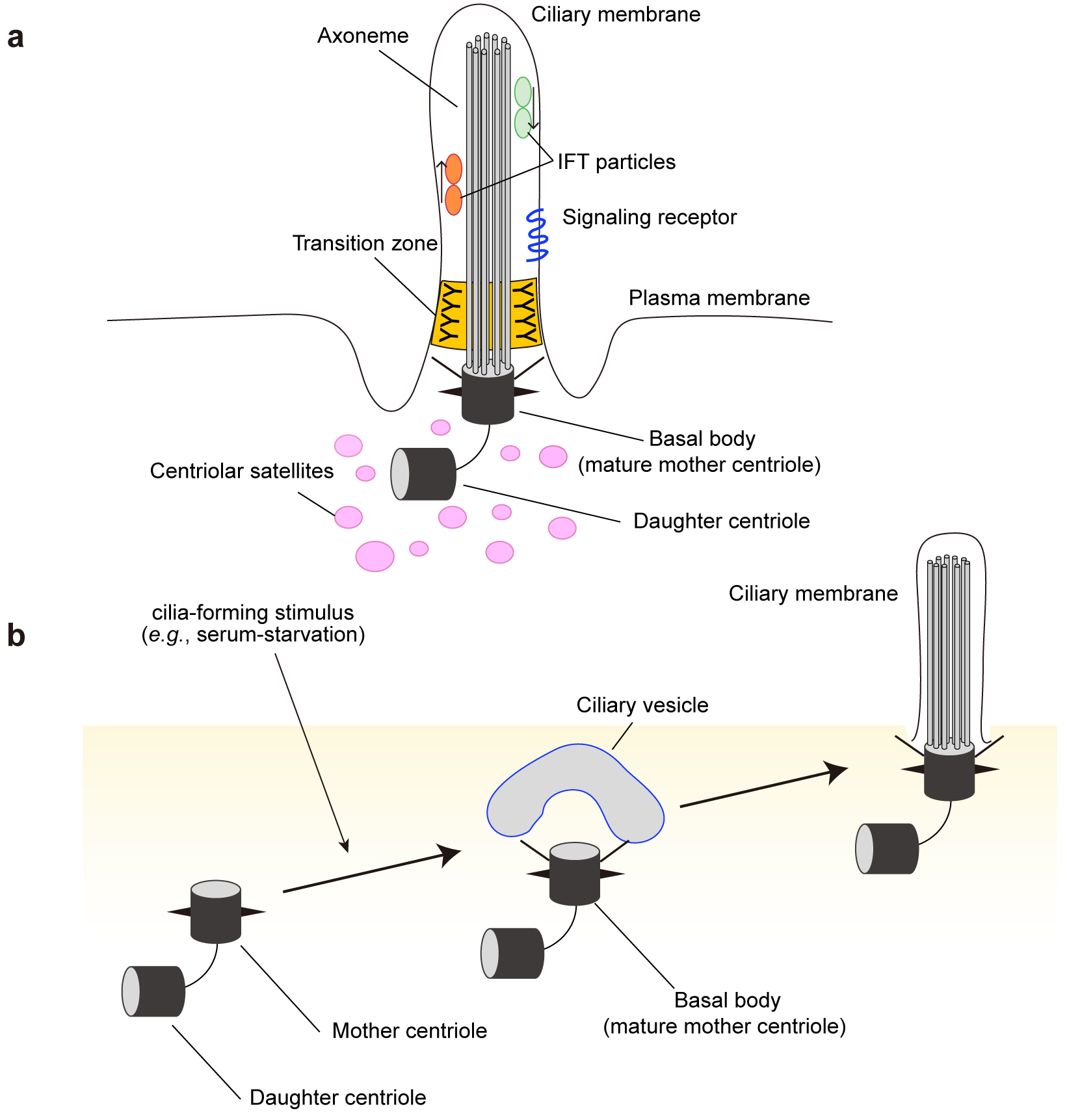
The primary cilium appears to sense, transduce, and integrate a variety of extracellular signals (16), (17), (18). Extracellular signals conveyed by primary cilia include mechanical stresses (bending of the cilium by fluid flow and tissue deformation), signaling proteins (e.g., Hedgehog [Hh], Wnt, Notch, platelet-derived growth factor, G protein-coupled receptors, growth factors, and hormones). In specialized cells, primary cilia can even respond to specific stimuli, including light, temperature, gravity, and osmolality (19). The Hh pathway is one of the most characterized cilium-dependent signaling pathways. It plays an essential role in regulating cell fate, stem cell renewal, and carcinogenesis, in addition to development and tissue homeostasis (20), (21). While the Hh pathway completely depends on primary cilia (22), other pathways, including the mTOR complex 1 (mTORC1) pathway, function through both cilium-dependent and -independent mechanisms. Signal transmission in the cilia regulates various physiological and developmental processes, such as embryonic patterning, tissue homeostasis, cell differentiation, and organogenesis (16), (19), (23).
Ciliary dysfunction leads to genetic syndromes known as ciliopathies. They present overlapping phenotypes including renal, hepatic and pancreatic cysts, skeletal defects, retinal degeneration, obesity, hearing loss, intellectual disability, and brain malformations (24), (25). Examples of ciliopathies include autosomal dominant and autosomal recessive polycystic kidney diseases, Bardet-Biedl syndrome (BBS), oral-facial-digital (OFD) type 1 syndrome, Joubert syndrome, nephronophthisis, Meckel-Grouber syndrome, and Birt-Hogg-Dubé (BHD) syndrome (24). In addition to these canonical ciliopathies, ciliogenesis appears to be compromised in various human cancers, including melanoma and pancreatic, breast, and renal cell cancers (26), (27).
Recent studies have shown that primary cilia, ciliary signaling pathways, and cilia-related proteins regulate autophagy and that, conversely, autophagy regulates ciliogenesis (28). In this review, we discuss recent findings on the crosstalk between cilia and autophagy. The possible involvement of cilia and autophagy in diseases is discussed in more detail in other studies (28).
While it has been known for a long time that serum starvation promotes ciliogenesis, the underlying mechanism remains unclear (29). Recent studies have discovered that autophagy, which is triggered in response to serum starvation, contributes to ciliogenesis by degrading specific proteins.
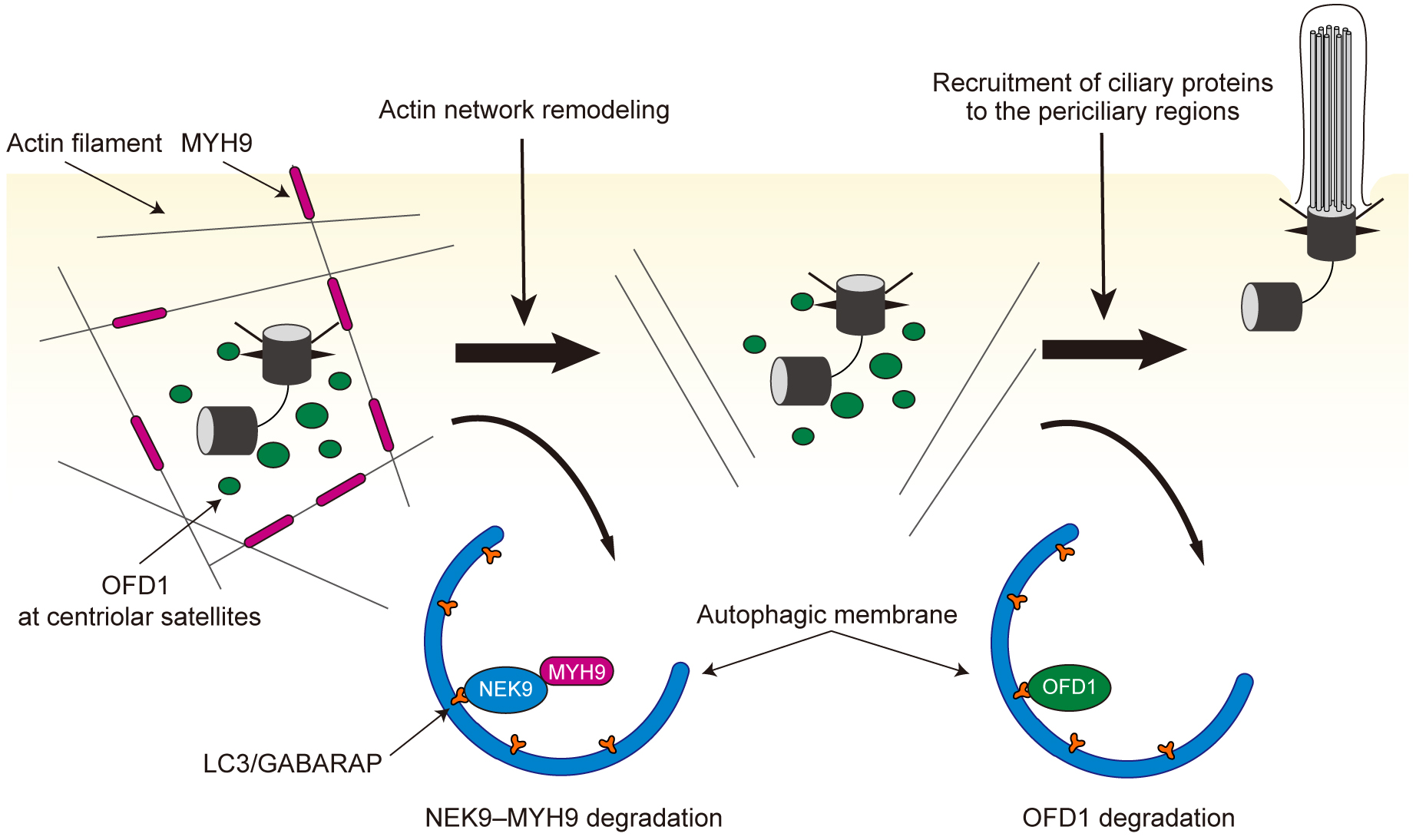
OFD1 was the first protein reported to be degraded by autophagy to promote ciliogenesis. Mutations in the OFD1 gene cause OFD syndrome (30). OFD1 localizes to the centriole and centriolar satellites that surround the centriole and has two opposing functions based on its localization (31), (32). OFD1 at the centriole acts as a promotor of ciliogenesis by facilitating migration of the basal body to the plasma membrane and allowing the recruitment of essential ciliary components such as IFT88 to the basal body (31), (33). Conversely, OFD1 at the centriolar satellites suppresses ciliogenesis by inhibiting the recruitment of ciliary proteins essential for cilia elongation such as BBS4. Under cilia-forming serum starvation conditions, autophagy degrades OFD1 at the centriolar satellites and promotes ciliogenesis (32). When autophagy is blocked, OFD1 accumulates at the centriolar satellites even in serum starvation conditions and impedes the recruitment of proteins required for cilia elongation, leading to impaired ciliogenesis. These results have led to the discovery of autophagy-mediated degradation of a cilia-related protein as a possibly important step in ciliogenesis. A recent study has shown that OFD1 has a LIR to interact with LC3, indicating that OFD1 at centriolar satellites is directly degraded by selective autophagy (34).
NIMA-related kinase 9 (NEK9) was also identified as a cilia-related protein that is degraded by selective autophagy (35). NEK9 has a LIR to interact with LC3 and GABARAP family proteins, and associates with autophagic membranes in a LIR-dependent manner (35), (36). Ciliogenesis was found to be impaired in cells harboring a mutation in the LIR of NEK9. Similarly, primary cilia formation was affected in the kidneys of mice carrying the same mutation. NEK9 interacts with MYH9 (also known as myosin IIA), a negative regulator of ciliogenesis (37), and mediates autophagic degradation of MYH9; in other words, NEK9 functions as a selective autophagy adaptor for MYH9. MYH9 suppresses ciliogenesis by stabilizing the actin network and inhibiting actin remodeling, which is essential for the early phase of ciliogenesis(38), (39). Thus, autophagic degradation of NEK9-MYH9 is a prerequisite for the proper progression of the early phase of ciliogenesis.
Autophagic degradation of NEK9-MYH9 and OFD1 is independent; NEK9-MYH9 and OFD1 at centriolar satellites seem to inhibit different steps in ciliogenesis. These findings have revealed that autophagy could play key roles at multiple stages of cilia formation (Figure 3) (35). Consistent with these findings, upregulation of autophagy promotes ciliogenesis in cells maintained in various culture conditions (32), (40), (41), (42), (43). It remains to be investigated whether NEK9-MYH9 and OFD1 are modified to be subjected to autophagy upon cilia-forming stimuli. Post-translational modification around the LIR is a well-known mechanism that conditionally regulates the activity of selective autophagy by changing the binding affinity between ATG8 proteins and the LIR (7). Thus, degradation of these cilia-related proteins by selective autophagy may be similarly regulated by post-translational modifications. Very recently, it was reported that ATG16L1 interacts with IFT20 and regulates ciliogenesis (44). However, this function appears to be independent of canonical autophagy.
Conversely, some reports have demonstrated that autophagy could negatively regulate ciliogenesis (45), (46). Under normal nutrient-rich conditions, autophagy occurs at basal levels and prevents unwanted cilia formation during cell proliferation by degrading proteins that are essential for cilia formation and assembly (45), (47). Autophagy-related proteins could localize at cilia or periciliary regions (45).
These findings suggest that the effects of autophagy on ciliogenesis may vary depending on various experimental conditions in culture cells, and that evaluation by physiologically relevant methods such as using mouse models would be more important than using only culture cells. Because primary cilia formation is impaired in the kidney of autophagy-deficient mice such as Atg5−/−;NSE-Atg5 and Atg7flox/flox;PEPCK-Cre mice, it seems reasonable to assume that, in general, autophagy positively regulates cilia formation under physiological conditions in vivo (35), (42).
Although autophagy regulates ciliogenesis, the relationship between autophagy and cilia is not unidirectional; primary cilia can also regulate autophagic activity. Because autophagy is activated by suppression of mTORC1 activity, which is regulated by primary cilia, it is plausible that primary cilia regulate autophagy (Figure 4a) (48). Autophagy activation upon serum starvation requires the presence of functional primary cilia; culture cells with impaired ciliogenesis, for example IFT20- and IFT88-depleted cells, exhibit decreased autophagy upon serum removal (45).
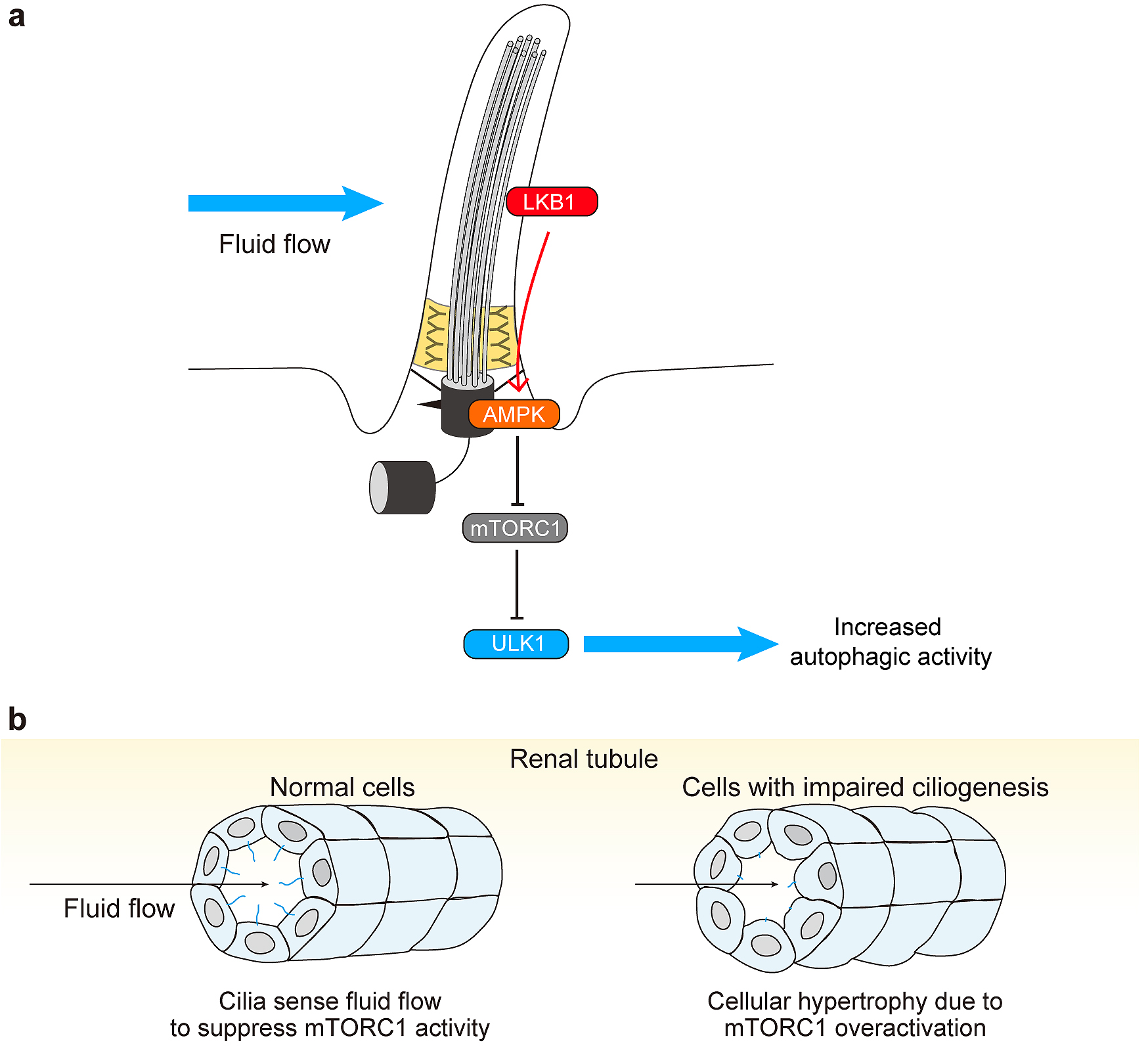
In normal ciliated cells, such as renal epithelial cells, primary cilia transduce extracellular shear stress induced by fluid flow and activate liver kinase B1 (LKB1), which localizes to the ciliary compartment and the basal body. LKB1 activates AMP-activated protein kinase localized at the basal body, which inactivates mTORC1 and activates the ULK1 kinase complex, leading to autophagy upregulation and cell size reduction (Figure 4a) (49), (50). Accordingly, cells with defective primary cilia show cellular hypertrophy (Figure 4b) (49), (51).
There is emerging evidence suggesting that primary cilium-dependent autophagy governs physiological adaptations in organs. A recent study revealed a key role for primary cilium-dependent autophagy in the eye (52). Induction of autophagy elicited by high intraocular pressure (IOP) and mechanical stretch of trabecular meshwork cells have been known to be essential in IOP (53). Shim et al. discovered that primary cilia act as a mechanosensor for mechanical stretch-induced autophagy and identified cross-regulatory talk between AKT1 and noncanonical SMAD2/3 signaling as critical elements of primary cilia-mediated autophagy activation by mechanical stretch (52). Removal of primary cilia disrupts the homeostatic IOP compensatory response and prevents autophagy induction in response to elevated pressure challenges, which supports the role of primary cilia-mediated autophagy in regulating IOP homeostasis.
In addition, another recent report has shown that shear stress caused by fluid flow stimulates lipophagy (selective degradation of lipid droplets by autophagy) in renal epithelial cells in a cilium-dependent manner (54). It facilitates the production of fatty acids that provide mitochondrial respiratory substrates through β-oxidation to generate ATP. Primary cilium-dependent lipophagy and mitochondrial biogenesis are required to support energy-consuming cellular processes that include glucose reabsorption, gluconeogenesis, and cytoskeletal remodeling. These findings reveal the involvement of primary cilia and autophagy in the transduction of extracellular mechanical forces into intracellular metabolic adaptation. The initiation of a specific type of autophagy by primary cilia in response to various extracellular stimuli (e.g., fluid flow, light, pressure, temperature, nutritional, and hormonal status) may be important for cells to adapt to different environments during differentiation and development. Indeed, another piece of evidence demonstrates that inhibition of cilia-mediated autophagy blocks proper neuroectodermal differentiation, suggesting a key role for primary cilium-dependent autophagy in differentiation (55).
Multiple studies have suggested that cilia-related proteins are directly involved in autophagy, not just in ciliogenesis. Inositol 5-phosphatase (INPP5E), a causative gene of Joubert syndrome, is a positive regulator of autophagosome maturation(56), (57). Initially, INPP5E was identified as a cilia-related protein that localizes to primary cilia. INPP5E depletion results in shorter cilia and affects transmission of Hh signaling (58), (59). A recent study has revealed that INPP5E mutations, which impair phosphatase activity, affect autophagic activity by disturbing autophagosome-lysosome fusion, suggesting the direct involvement of cilia-related protein in autophagy (57). In addition, another cilia-related protein called inositol phosphate-5-phosphatase, known as OCRL, which localizes to basal bodies and cilia and positively regulates ciliogenesis, also plays a functional role in autophagy (60), (61). OCRL is a causative gene for Lowe syndrome and Dent-2 disease, in which fibroblasts present impaired ciliogenesis (62). Similar to INPP5E, OCRL also regulates autophagosome-lysosome fusion; loss of catalytic activity of OCRL causes autophagosome accumulation and lysosomal dysfunction in cells isolated from patients with Lowe syndrome (61).
Folliculin (FLCN) localizes to primary cilia and is essential for ciliogenesis and cilium-dependent signaling such as the Wnt and planar cell polarity pathways (63). Mutations in the FLCN gene cause BHD syndrome (64). In addition to its ciliary function, FLCN also localizes to lysosomes and acts as an activator of RagC/D GTPases (65). Kidney samples from patients with BHD show autophagic defects (66). A recent study indicated that FLCN is crucial for mTORC1-mediated phosphorylation of transcription factor EB (TFEB), a master regulator of lysosomal biogenesis and autophagy, and that abnormal constitutive TFEB activation is the main driver of kidney abnormalities (e.g., renal cysts and renal carcinoma) and mTORC1 hyperactivity in a mouse model of BHD syndrome (67). Whether overactivation of TFEB is also involved in other clinical manifestations of BHD syndrome is a subject of future research, but some of ciliopathy-related symptoms of BHD may be primarily due to abnormal activation of mTOR and suppression of autophagy rather than to impaired ciliogenesis.
We have described the bidirectional relationship between autophagy and the primary cilium; autophagy regulates ciliogenesis positively or negatively, likely depending on the cellular context, and primary cilia regulate autophagy. Although there has been significant progress during the past decade, there remain further questions. For example, both autophagy and ciliogenesis are stimulated by serum starvation, but it is unclear whether this is merely a coincidence or whether there is a causal relationship. Amino acid starvation induces autophagic degradation of OFD1 and NEK9-MYH9, but not ciliogenesis, indicating that the degradation of these factors is not sufficient for ciliogenesis and signals triggered by serum starvation would be crucial. It would be valuable to determine whether it is important to achieve acute reduction of OFD1 and MYH9 during ciliogenesis or to keep their basal levels low. Also, there is almost nothing known about the relationship between autophagy and motile cilia. Further studies will expand our knowledge of the cilia-autophagy axis, which is vital for cellular and organ homeostasis.
This article is based on the study, which received the Medical Award of The Japan Medical Association in 2020.
None
This work was supported by Exploratory Research for Advanced Technology (ERATO) (No. JPMJER1702 to N.M.) from the Japan Science and Technology Agency (JST).
Y.Y. and N.M. wrote the manuscript.
Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564-76.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349-64.
Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020;21(8):439-58.
Søreng K, Neufeld TP, Simonsen A. Membrane trafficking in autophagy. Int Rev Cell Mol Biol. 2018;336:1-92.
Saxton RA, Sabatini DM. MTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960-76.
Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233-42.
Johansen T, Lamark T. Selective autophagy: ATG8 family proteinS, LIR motifs and cargo receptors. J Mol Biol. 2020;432(1):80-103.
Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014;54(2):224-33.
Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013-22.
Chino H, Mizushima N. ER-Phagy: quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020;30(5):384-98.
Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017;41(1):10-22.
Papadopoulos C, Kravic B, Meyer H. Repair or lysophagy: dealing with damaged lysosomes. J Mol Biol. 2020;432(1):231-9.
Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2020;63:1-10.
Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif―crucial for selective autophagy. J Cell Sci. 2013;126(15):3237-47.
Malicki JJ, Johnson CA. The cilium: cellular antenna and Central Processing Unit. Trends Cell Biol. 2017;27(2):126-40.
Anvarian Z, Mykytyn K, Mukhopadhyay S, et al. Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol. 2019;15(4):199-219.
Spasic M, Jacobs CR. Primary cilia: cell and molecular mechanosensors directing whole tissue function. Semin Cell Dev Biol. 2017;71:42-52.
Berbari NF, O’Connor AK, Haycraft CJ, et al. The primary cilium as a complex signaling center. Curr Biol. 2009;19(13):R526-35.
Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol. 2018;6:8.
Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406-10.
Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445-57.
Bangs F, Anderson KV. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol. 2017;9(5).
Tummala P, Arnsdorf EJ, Jacobs CR. The role of primary cilia in mesenchymal stem cell differentiation: A pivotal switch in guiding lineage commitment. Cell Mol Bioeng. 2010;3(3):207-12.
Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533-47.
Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533-43.
Lee J, Yi S, Kang YE, et al. Defective ciliogenesis in thyroid hurthle cell tumors is associated with increased autophagy. Oncotarget. 2016;7(48):79117-30.
Cao M, Zhong Q. Cilia in autophagy and cancer. Cilia. 2015;5:4.
Morleo M, Franco B. The autophagy-Cilia axis: an intricate relationship. Cells. 2019;8(8):905.
Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979;17(3):527-35.
Ferrante MI, Giorgio G, Feather SA, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569-76.
Ferrante MI, Zullo A, Barra A, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112-7.
Tang Z, Lin MG, Stowe TR, et al. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502(7470):254-7.
Singla V, Romaguera-Ros M, Garcia-Verdugo JM, et al. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell. 2010;18(3):410-24.
Morleo M, Brillante S, Formisano U, et al. Regulation of autophagosome biogenesis by OFD1-mediated selective autophagy. EMBO J. 2021;40(4):e105120.
Yamamoto Y, Chino H, Tsukamoto S, et al. NEK9 regulates primary cilia formation by acting as a selective autophagy adaptor for MYH9/myosin IIA. Nat Commun. Forthcoming 2021.
Shrestha BK, Skytte Rasmussen M, Abudu YP, et al. NIMA-related kinase 9-mediated phosphorylation of the microtubule-associated LC3B protein at Thr-50 suppresses selective autophagy of p62/sequestosome 1. J Biol Chem. 2020;295(5):1240-60.
Pitaval A, Senger F, Letort G, et al. Microtubule stabilization drives 3D centrosome migration to initiate primary ciliogenesis. J Cell Biol. 2017;216(11):3713-28.
Copeland J. Actin-based regulation of ciliogenesis―the long and the short of it. Semin Cell Dev Biol. 2020;102:132-8.
Kim J, Jo H, Hong H, et al. Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat Commun. 2015;6:6781.
Liu ZQ, Lee JN, Son M, et al. Ciliogenesis is reciprocally regulated by PPARA and NR1H4/FXR through controlling autophagy in vitro and in vivo. Autophagy. 2018;14(6):1011-27.
Hsiao CJ, Chang CH, Ibrahim RB, et al. Gli2 modulates cell cycle re-entry through autophagy-mediated regulation of the length of primary cilia. J Cell Sci. 2018;131(24).
Wang S, Livingston MJ, Su Y, et al. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy. 2015;11(4):607-16.
Kim ES, Shin JH, Park SJ, et al. Inhibition of autophagy suppresses sertraline-mediated primary ciliogenesis in retinal pigment epithelium cells. PLOS ONE. 2015;10(2):e0118190.
Boukhalfa A, Roccio F, Dupont N, et al. The autophagy protein ATG16L1 cooperates with IFT20 and INPP5E to regulate the turnover of phosphoinositides at the primary cilium. Cell Rep. 2021;35(4):109045.
Pampliega O, Orhon I, Patel B, et al. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502(7470):194-200.
Struchtrup A, Wiegering A, Stork B, et al. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy. 2018;14(4):567-83.
Follit JA, Tuft RA, Fogarty KE, et al. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell. 2006;17(9):3781-92.
Lai Y, Jiang Y. Reciprocal regulation between primary cilia and mTORC1. Genes (Basel). 2020;11(6):711.
Orhon I, Dupont N, Zaidan M, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016;18(6):657-67.
Zhong M, Zhao X, Li J, et al. Tumor suppressor folliculin regulates mTORC1 through primary cilia. J Biol Chem. 2016;291(22):11689-97.
Boehlke C, Kotsis F, Patel V, et al. Primary cilia regulate mTORC1 activity and cell size through LKB1. Nat Cell Biol. 2010;12(11):1115-22.
Shim MS, Nettesheim A, Dixon A, et al. Primary cilia and the reciprocal activation of AKT and SMAD2/3 regulate stretch-induced autophagy in trabecular meshwork cells. Proc Natl Acad Sci U S A. 2021;118(13):e2021942118.
Porter KM, Jeyabalan N, Liton PB. MTOR-independent induction of autophagy in trabecular meshwork cells subjected to biaxial stretch. Biochim Biophys Acta. 2014;1843(6):1054-62.
Miceli C, Roccio F, Penalva-Mousset L, et al. The primary cilium and lipophagy translate mechanical forces to direct metabolic adaptation of kidney epithelial cells. Nat Cell Biol. 2020;22(9):1091-102.
Jang J, Wang Y, Lalli MA, et al. Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell. 2016;165(2):410-20.
Bielas SL, Silhavy JL, Brancati F, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41(9):1032-6.
Hasegawa J, Iwamoto R, Otomo T, et al. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J. 2016;35(17):1853-67.
Chávez M, Ena S, Van Sande J, et al. Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev Cell. 2015;34(3):338-50.
Garcia-Gonzalo FR, Phua SC, Roberson EC, et al. Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev Cell. 2015;34(4):400-9.
Luo N, West CC, Murga-Zamalloa CA, et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet. 2012;21(15):3333-44.
De Leo MG, Staiano L, Vicinanza M, et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol. 2016;18(8):839-50.
Coon BG, Hernandez V, Madhivanan K, et al. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet. 2012;21(8):1835-47.
Luijten MN, Basten SG, Claessens T, et al. Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum Mol Genet. 2013;22(21):4383-97.
Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2(2):157-64.
Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429-33.
Dunlop EA, Seifan S, Claessens T, et al. FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy. 2014;10(10):1749-60.
Napolitano G, Di Malta C, Esposito A, et al. A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature. 2020;585(7826):597-602.
