Corresponding author: Yoshishige Miyabe, y-miyabe@nms.ac.jp
DOI: 10.31662/jmaj.2020-0019
Received: March 30, 2020
Accepted: April 27, 2020
Advance Publication: July 13, 2020
Published: July 15, 2020
Cite this article as:
Miyabe Y, Miyabe C, Iwai Y, Luster AD. Targeting the Chemokine System in Rheumatoid Arthritis and Vasculitis. JMA J. 2020;3(3):182-192.
Arrest of circulating leukocytes and subsequent diapedesis is a fundamental component of inflammation. In general, the leukocyte migration cascade is tightly regulated by chemoattractants, such as chemokines. Chemokines, small secreted chemotactic cytokines, as well as their G-protein-coupled seven transmembrane spanning receptors, control the migratory patterns, positioning and cellular interactions of immune cells. Increased levels of chemokines and their receptors are found in the blood and within inflamed tissue in patients with rheumatoid arthritis (RA) and vasculitis. Chemokine ligand-receptor interactions regulate the recruitment of leukocytes into tissue, thus contributing in important ways to the pathogenesis of RA and vasculitis. Despite the fact that blockade of chemokines and chemokine receptors in animal models have yielded promising results, human clinical trials in RA using inhibitors of chemokines and their receptors have generally failed to show clinical benefits. However, recent early phase clinical trials suggest that strategies blocking specific chemokines may have clinical benefits in RA, demonstrating that the chemokine system remains a promising therapeutic target for rheumatic diseases, such as RA and vasuculitis and requires further study.
Key words: Chemokine, Rheumatoid arthritis, Vasculitis
Egress of leukocytes from the circulation into the extravascular space is a fundamental component of inflammation. Leukocyte recruitment from blood into tissues follows a well-established paradigm: (i) tethering and rolling on the vessel wall, (ii) firm arrest on the endothelium, (iii) spreading out and crawling in all directions on the vessel, and transendothelial migration to extravasate into tissue. This process is tightly controlled by adhesion molecules and chemoattractants including chemokines (1).
Chemokines are chemotactic cytokines that regulate the migration of a wide variety of immune cells (2). In the context of the immune system, chemokines control the positioning of all immune cells and contribute to immune cell development, homeostasis and inflammation. More than 50 chemokines and 19 chemokine receptors have been identified in humans and mice (Table 1) (2). These small (8-10 kDa) secreted proteins are classified into four groups on the location of cysteine residues near the N terminus of their primary amino acid sequence: XC-chemokines contain a single N-terminal cysteine, CC-chemokines have two adjacent residues, CXC-chemokines have two cysteines separated by one other amino acid, and CX3C-chemokines have three amino acids between the cysteine residues (2). Most chemokines are secreted into the extracellular space where they bind heparin-like glycosaminoglycans on the cell surface and are embedded in the extracellular matrix, forming transient or stable gradients. Chemokines and their gradients are detected by binding to specific chemokine receptors. Chemokine receptors are seven-transmembrane receptors and are expressed on the surface of immune cells. These receptors are G-protein coupled receptors that signal via Gi-type G proteins. There are approximately 20 classical chemokine receptors coupled to G proteins and regulate cell migration, whereas 5 atypical chemokine receptors that do not couple to G proteins and do not induce cell migration have been identified (Table 2) (2).
Table 1. Chemokines and Their Receptors.
| Chemokine | Other names | Receptor |
|---|---|---|
| CXCL1 | GROα, MGSA, mouse KC | CXCR2, ACKR1 |
| CXCL2 | GROβ, MIP-2α, mouse MIP2 | CXCR2, ACKR1 |
| CXCL3 | GROγ, MIP-2β | CXCR2, ACKR1 |
| CXCL4 | PF4 | Unknown |
| CXCL4L1 | PF4V1 | Unknown |
| CXCL5 | ENA-78, mouse LIXa | CXCR2, ACKR1 |
| CXCL6 | GCP-2 (human only) | CXCR1, CXCR2, ACKR1 |
| CXCL7 | NAP-2 | CXCR2, ACKR1 |
| CXCL8 | IL-8 (human only) | CXCR1, CXCR2, ACKR1 |
| CXCL9 | Mig | CXCR3 |
| CXCL10 | IP-10 | CXCR3 |
| CXCL11 | I-TAC | CXCR3, ACKR1, ACKR3 |
| CXCL12 | SDF-1 | CXCR4, ACKR3 |
| CXCL13 | BLC, BCA-1 | CXCR5, ACKR1, ACKR4 |
| CXCL14 | BRAK | Unknown |
| CXCL15 | Lungkine (mouse only) | Unknown |
| CXCL16 | CXCR6 | |
| CCL1 | I-309, mouse TCA3 | CCR8 |
| CCL2 | MCP-1, mouse JE | CCR2, ACKR1, ACKR2 |
| CCL3b | MIP-1α, LD78α | CCR1, CCR5, ACKR2 |
| CCL3L1 | LD78β | CCR1, CCR3, CCR5, ACKR2 |
| CCL4 | MIP-1β | CCR5, ACKR2 |
| CCL4L1 | LAG-1 | CCR5 |
| CCL5 | RANTES | CCR1, CCR3, CCR5, ACKR2 |
| CCL6 | C-10, MRP-1 (mouse only) | Unknown |
| CCL7 | MCP-3, mouse Fic or MARC | CCR2, CCR3, ACKR1, ACKR2 |
| CCL8 | MCP-2 | Human: CCR1, CCR2, CCR3, CCR5, ACKR1, ACKR2; mouse: CCR8, ACKR1, ACKR2 |
| CCL9/10 | MIP-1γ, MRP-2 (mouse only) | Unknown |
| CCL11 | Eotaxin-1 | CCR3, ACKR2 |
| CCL12 | MCP-5 (mouse only) | CCR2 |
| CCL13 | MCP-4 (human only) | CCR2, CCR3, CCR5, ACKR1, ACKR2 |
| CCL14 | HCC-1 (human only) | CCR1, ACKR1, ACKR2 |
| CCL15 | Leukotactin-1, HCC-2, MIP-5 (human only) | CCR1, CCR3 |
| CCL16 | HCC-4, NCC-4, LEC (human only) | CCR1, CCR2, CCR5, ACKR1 |
| CCL17 | TARC | CCR4, ACKR1, ACKR2 |
| CCL18 | PARC, DC-CK1 (human only) | CCR8 |
| CCL19 | MIP-3β, ELC | CCR7, ACKR4 |
| CCL20 | MIP-3α, LARC | CCR6 |
| CCL21 | SLC, 6CKine | CCR6, CCR7, ACKR4 |
| CCL22 | MDC | CCR4, ACKR1, ACKR2 |
| CCL23 | MPIF-1, MMP-3 (human only) | Unknown |
| CCL24 | Eotaxin-2, MPIF-2 | CCR3 |
| CCL25 | TECK | CCR9, ACKR4 |
| CCL26 | Eotaxin-3 | CCR3, CX3CR1 |
| CCL27 | CTAK | CCR10 |
| CCL28 | MEC | CCR3, CCR10 |
| XCL1 | Lymphotactin α, SCM-1α | XCR1 |
| XCL2 | Lymphotactin β, SCM-1β | XCR1 |
| CX3CL1 | Fractalkine | CX3CR1 |
Table 2. Chemokine Receptors: Cellular Expression and Primary Function.
| Receptor | Cell expression | General immune function |
|---|---|---|
| Classical chemokine receptors | ||
| CXCR1 | Neutrophils and lower expression on monocytes, NK cells, mast cells, basophils, CD8+ TEFF cells and endothelial cells | Neutrophil trafficking |
| CXCR2 | Neutrophils and lower expression on monocytes, NK cells, mast cells, basophils, CD8+ T cells and endothelial cells | B cell lymphopoiesis, neutrophil egress from bone marrow and neutrophil trafficking |
| CXCR3 | TH1 cells, CD8+TCM cells and TEM cells, NK cells, NKT cells, plasmacytoid dendritic cells, B cells, Treg cells and TFH cells | Type 1 adaptive immunity |
| CXCR4 | Most leukocytes and endothelial cells | Hematopoiesis, organogenesis and bone marrow homing |
| CXCR5 | B cells, TFH cells, TFR cells CD8+ TEM cells | B cell and T cell trafficking in lymphoid tissue to the B cell zone or follicles |
| CXCR6 | TH1 cells, TH17 cells, γδT cells, ILCs, NKT cells, NK cells, plasma cells, endothelial cells | ILC function and adaptive immunity |
| CCR1 | Monocytes, macrophages, neutrophils, TH1 cells, basophils, dendritic cells | Innate immunity and adaptive immunity |
| CCR2 | Monocytes, macrophages, TH1 cells, immature dendritic cells, basophils, NK cells and endothelial cells | Monocyte trafficking and type 1 adaptive immunity |
| CCR3 | Eosinophils and lower expression on basophils and mast cells | Type 2 adaptive immunity, eosinophil distribution and trafficking |
| CCR4 | TH2 cells, skin-homing T cells and lung-homing T cells, Treg cells and lower expression on TH17 cells, CD8+T cells, monocytes, B cells and immature dendritic cells | Homing of T cells to the skin and lungs and type 2 immune responses |
| CCR5 | Monocytes, macrophages, TH1 cells, NK cells, Treg cells, CD8+ T cells, dendritic cells and neutrophils | Type 1 adaptive immunity |
| CCR6 | TH17 cells, immature dendritic cells, γδT cells, NKT cells, NK cells, Treg cells and TFH cells | Immature dendritic cell trafficking, GALT development and type 17 adaptive immune responses |
| CCR7 | Naïve T cells, TCM cells, TRCM cells, mature dendritic, B cells | Mature dendritic cell, B cell and T cell trafficking in lymphoid tissue to T cell zone and egress of dendritic cells and T cells from tissue |
| CCR8 | TH2 cells, Treg cells, skin TRM cells, γδT cells, monocytes and macrophage | Immune surveillance in skin, type 2 adaptive immunity and thymopoiesis |
| CCR9 | Gut-homing T cells, thymocytes, B cells, dendritic cells and plasmacytoid dendritic cells | Homing of T cells to the gut, GALT development and function and thymopoiesis |
| CCR10 | Skin-homing T cells and IgA-plasmablasts | Homing immunity at mucosal sites and immune surveillance in the skin |
| XCR1 | Cross-presenting CD8+ dendritic cells and thymic dendritic cells | Antigen cross-presentation by CD8+ dendritic cells |
| CX3CR1 | Resident monocytes, macrophages, microglia, TH1 cells, CD8+ TEM cells, NK cells, γδT cells and dendritic cells | Patrolling monocytes in innate immunity, microglial cell and NK cell migration and type 1 adaptive immunity |
| Atypical chemokine receptors | ||
| ACKR1 (DARC) | Red blood cells and endothelial cells | Chemokine transcytosis and chemokine scavenging |
| ACKR2 (D6) | Dendritic cells, B cells and lymphatic endothelium | Chemokine scavenging |
| ACKR3 (CXCR7) | B cell and stromal cells | Shaping chemokine gradients for CXCR4 |
| ACKR4 (CCRL1; CCX-CKR) | Thymic epithelium | Chemokine scavenging |
| DARC, Duffy Antigen Receptor for Chemokines; GALT, gut-associated lymphoid tissue; ILCs, innate lymphoid cells; NK, natural killer; NKT, natural killer T; TCM cell, central memory T cell; TEFF cell, effector T cell; TEM cell, effector-memory T cell; TFH cell, follicular helper T cell; TFR cell, follicular regulatory T cell; TH cell, T helper; TRCM, recirculating memory T cell; TREG cell, regulatory T cell; TRM cell, resident-memory T cell. | ||
The chemokine system and other inflammatory mediators might play a central role in chronic inflammation such as autoimmune diseases. In fact, high levels of several chemokines are seen in the blood and disease tissues, such as the joints and blood vessels, in patients with rheumatoid arthritis (RA) (3) and vasculitis (4), and their levels have been associated with the disease severity and/or activity. Chemokines and their receptors likely contribute to the recruitment of immune cells into the affected tissue in RA and vasculitis and are also thought to contribute to the activation of leukocytes once in the tissue, resulting in integrin activation and the production of proteases and inflammatory mediators (5). Thus, the control of leukocyte entry into the tissue represents a major point at which new therapeutics, such as blockade of the chemokine system, could be developed to attenuate inflammation in RA and vasculitis.
In this review, we summarize the pathogenic roles of chemokines and their receptors in RA and vasculitis. Additionally, we provide an update on the clinical trials of chemokine- and chemokine-receptor-targeting drugs in arthritis and vasculitis and discuss their potential as therapeutic targets.
Inflammatory arthritis, including rheumatoid arthritis (RA), is characterized by immune cell infiltration into joints, resulting in pannus formation with associated cartilage and bone destruction. This immune cell infiltrate drives RA pathogenesis and is composed of multiple cell types, including lymphocytes, macrophages, and neutrophils. Approximately 1 million people in Japan have RA. If not effectively treated, RA causes irreversible joint destruction resulting in significant disability, increased risk of cardiovascular disease, and markedly increased medical costs. Biological therapies, such as TNF inhibitors and IL-6 inhibitors, have revolutionized the treatment of RA. However, ~50% of RA patients still do not respond to these agents, and patients who do respond often have a residual disease and are at an increased risk of infection, such as pneumonia, associated with their use (6). Therefore, there remains an ongoing need to identify new therapeutic targets and treatment strategies for RA.
Animal models of inflammatory arthritis have provided valuable research tools for understanding the pathogenic mechanism of RA and for studying therapeutic targets. In fact, several animal models of inflammatory arthritis, such as the K/BxN arthritogenic serum transfer model of arthritis (7), the type II collagen-induced arthritis (CIA) (8) and the collagen antibody-induced arthritis (CAIA) (9), have a number of similarities to RA, including synovial hyperplasia, mononuclear cell infiltration and bone destruction. Therefore, these animal models have been used to define the role of chemokines and their receptors in inflammatory arthritis. In this section, we describe the functional role of chemokines and their receptors in animal models and in human RA and discuss therapies targeting these molecules.
Numerous studies have demonstrated that multiple chemokines are highly expressed in serum, synovial fluid and synovial tissue of patients with RA, compared to healthy controls. For instance, the CXC-chemokines (CXCL5, CXCL8, CXCL9, CXCL10, CXCL12, CXCL13 and CXCL16), the CC-chemokines (CCL2, CCL3, CCL4, CCL5, CCL18, CCL19, CCL20, CCL21, CCL25), and the CX3C-chemokine CX3CL1 are increased in serum, synovial fluid, and synovial tissue of RA patients, compared with healthy controls (10), (11), (12), (13), (14), (15). In the pathogenesis of human RA, CXC-chemokines promote neutrophils (CXCL1, CXCL2, CXCL5 and CXCL8) (16), the effector T cell (CXCL10) (17) and the B cell (CXCL13) (18), (19) recruitment into the joint. CC-chemokines induce monocytes (CCL2, CCL3, CCL4, CCL5 and CCL7) (20), (21), T cells (CCL18, CCL19, CCL20, CCL21 and CCL25) (22) and the B cell (CCL20) (23) entry into the inflamed joint, whereas CX3CL1 induces monocyte recruitment (24).
In addition, CXCL8, CXCL12 and CXCL16 induce angiogenesis in synovial tissue in RA, and CXCL8, CXCL10 and CXCL13 are promising biomarkers of RA disease activity/severity (11), (25). Synovial macrophages and fibroblast-like synoviocytes (FLSs) might be the main producers of many inflammatory chemokines in the joints of RA patients, whereas synovial endothelial cells mainly produce CX3CL1 and some CC-chemokines in RA patients. However, the pathogenic roles of chemokines in human RA remain unclear.
These same chemokines are also elevated in the serum, synovial fluid and synovial tissue in animal models of inflammatory arthritis, compared with control animals (20), (21), (22), (24), (26), (27), (28), (29), (30), (31), (32). Thus, animal models of arthritis are available to dissect the pathogenic roles of chemokines in inflammatory arthritis. Several studies using the animal models have indicated the development of arthritis in RA: (i) immune complexes stimulate synovial macrophages to generate CXCL1 that initiates neutrophil recruitment into the joints in the early phase of animal models of arthritis (33); (ii) after neutrophils enter the inflamed tissue, they become activated and produce CXCL2, which contributes to the recruitment of additional neutrophils in a positive-feedback loop (26), (33); (iii) neutrophils can also amplify the local response by secreting inflammatory cytokines, such as IL-1, that induce chemokine production by FLSs in the joint (26); (iv) In later phases of the disease, FLSs and other cells might play important roles in recruiting T and B cells (Figure 1).
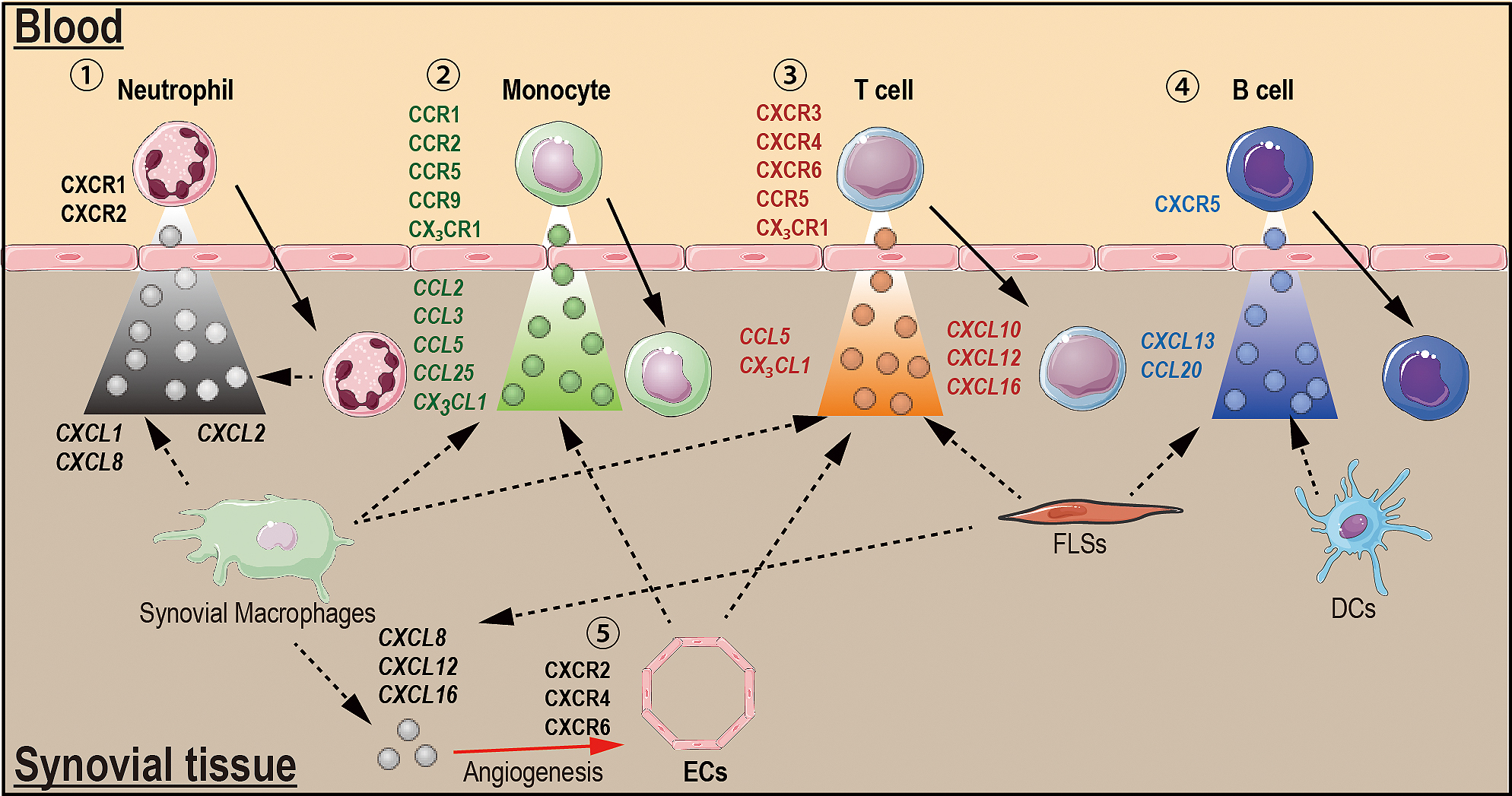
Multiple chemokine receptors (CXCR1-6, CCR1, CCR2, CCR5-7, CCR9 and CCR10) and chemokines are all highly expressed in the joints of human RA and in animal models of arthritis (10), (20), (22), (26), (27), (34), (35). In addition, chemokine receptors control the recruitment of neutrophils (CXCR1 and CXCR2), T helper type 1 (TH1) cell (CXCR3), lymphocyte (CXCR4), B cell (CXCR5), TFH cells (CXCR5), T cell (CXCR6, CCR5 and CCR6), monocyte (CCR1, CCR2, CCR5-7, CCR9 and CCR10) and DCs (CCR9) into the inflamed joints in human RA and in animal models (Figure 1) (10), (17), (18), (19), (20), (22), (23), (26), (35), (36). The CX3C-chemokine receptor CX3CR1 is expressed on CD3-positive T cells in the RA synovium (15), suggesting that the receptor also contributes to the recruitment of T cells into RA joints (Figure 1). Thus, different combinations of chemokine receptors are required for the entry of different cell types into the synovium during RA. For instance, CXCR1 and CXCR2 are required for neutrophils, CCR2, CCR5, CCR9, and CX3CR1 for monocytes; CXCR3, CXCR4, CXCR6, CCR5, CCR6 and CX3CR1 for T cells; and CXCR5 for B cells (Figure 1).
Some chemokine receptors are also expressed on stromal cells in RA patients, although their functions on these cells are largely unknown. Although chemokine receptor signaling in synovial endothelial cells (ECs) during RA may be an important mediator of angiogenesis, others in RA FLSs may contribute to induce the production of inflammatory mediators. For instance, CCL21/CCR7 and CCL28/CCR10 signaling in synovial ECs promotes RA angiogenesis (37), (38). CCR9, which is highly expressed on FLSs during RA, induces the production of inflammatory cytokines in vitro upon stimulation with CCL25, although the role of this interaction in vivo is not yet clear (22). These studies suggest that in addition to immune cells, the chemokine system also plays important roles in the function of stromal cells, such as ECs and FLSs, in the pathogenesis of RA.
Atypical chemokine receptors (ACKRs) are homologous to classical chemokine receptors and bind chemokines but do not couple to G proteins. These atypical receptors appear to play a primary role in shaping chemokine gradients by scavenging and controlling transcytosis of chemokines. Atypical chemokine receptor 1 (ACKR1/DARC) and ACKR3/CXCR7 are expressed on joint ECs in human RA synovial tissue (13), (39), (40). It has recently been reported that ACKR2/D6 expression is elevated on peripheral blood cells and on leukocytes and stromal cells in synovial tissue of RA patients (41). More recently, we have demonstrated that ACKR1/DARC has transcytosed inflammatory chemokines (CXCL1 and CXCL2) to initiate neutrophil entry into the joint in animal models of inflammatory arthritis (39), (42). Meanwhile, ACKR3/CXCR7 has been demonstrated to play a role in angiogenesis in an animal model of arthritis (13). However, the functional role of ACKRs in human RA pathogenesis remains unknown.
Numerous chemokines and their receptor inhibitors have been investigated and tested in the animal models. For instance, blockade of a single chemokine (CXCL10, CXCL13, CCL2, CX3CL1 or XCL1) or chemokine receptors (CXCR2, CXCR3, CXCR5, CXCR7, CCR1, CCR2, CCR5, CCR7 and CCR9) have demonstrated preventive and, in some cases, therapeutic efficacy in animal models (13), (17), (21), (22), (24), (26), (27), (36), (43), (44), (45), (46), (47), (48), (49), (50), (51), (52), (53), (54). However, only a few clinical trials (CXCL10; MDX-10 (14), CX3CL1; E6011 and CCR1; CCX354-C (55)) of 10 drugs that target the chemokine system had some clinical benefits in RA patients (Table 3).
Table 3. Targeting the Chemokine Systems in Rheumatoid Arthritis.
| Target | Drug (Type of drug) | Type of study | Efficacy | Study outcome |
|---|---|---|---|---|
| CXCL10 | MDX-1100 (antibody) | Phase II | Mildly effective | The ACR20 response at week 12 was 54% (MDX1100 and MTX) and 17% (placebo and MTX) |
| CCL2 | ABN912 (antibody) | Phase Ib | Not effective | ABN912 did not result in any clinical improvement. |
| CCR1 | CP-481,715 (small molecules) | Phase Ib | Mildly effective | CP-481,715 reduced tender and swollen joint count, and macrophages infiltration into the synovial tissue than those of placebo. |
| CCX354-C (small molecules) | Phase II | Mildly effective | The ACR20 response at week 12 was 39% (placebo), 43% (CCX354-C; 100mg twice daily) and 52% (CCX354-C; 200 mg once daily) | |
| MLN3897 (small molecules) | Phase IIa | Not effective | The ACR20 response at week 12 was 35% (MLN3897) and 33% (placebo). | |
| CCR2 | MLN1202 (antibody) | Phase IIa | Not effective | Patients treated with CCR2 monoclonal antibody or placebo for 6 weeks. No clinical improvement |
| CCR5 | SCH351125 (small molecules) | Phase Ib | Not effective | The ACR20 response at week 4 was 20% (SCH351125) and 33% (placebo). |
| AZD5672 (small molecules) | Phase IIb | Not effective | The ACR20 response at week 12 was around 35% (AZD5672) and 38% (placebo). | |
| UK-427,857 (small molecules) | Phase IIa | Not effective | The ACR20 response at week 12 was 23.7% (UK-427,857) and 23.8% (placebo). | |
| CX3CL1 | E6011 (antibody) | Phase I/II | Effective? (no placebo) | ~60% treated patients had at ACR20 response at week 12. |
| This table is modified from Miyabe Y, Lian J, Miyabe C, et al. Chemokines in rheumatic diseases: pathogenic role and therapeutic implications. Nat Rev Rheumatol. 2019;15(12):731-46 (5). The authors have the right to use the original table in Reference 5 and got the permission from Springer Nature. | ||||
Despite many inhibitors targeting the chemokine system failed to show positive results in clinical trials, indicating that the chemokine system might not be therapeutic targets for RA may be premature. CCR1 inhibitors might be an instructive example. MLN3897 was found to have a much lower pharmacokinetic and pharmacodynamic profile for CCR1 blockade compared to CCX354-C (56), suggesting that the lack of success of MLN3897 was not necessarily due to CCR1 being a poor target in terms of its underlying biology. Thus, CP-481,715 and MLN3897 failed to show any beneficial effects in clinical trials (Table 3) (55). It will be also important for inhibitors targeting the chemokine system to determine if optimum pharmacokinetic properties and receptor inhibition have been achieved before making final conclusions regarding the usefulness of inhibiting that particular target in a given disease process.
In addition, a more recent study of the animal model demonstrated that broadly cross-reactive chemokine-blocking antibodies for CXCR2 ligands (CXCL1-3 and CXCL5) dramatically attenuated inflammatory arthritis compared with blocking antibodies against a single chemokine (CXCL1) (57). Because the functions, such as adhesion and extravasation, during leukocyte trafficking overlaps between many chemokine systems, inhibition of a single chemokine system alone might not be sufficient to completely suppress leukocyte recruitment. Moreover, the pathogenesis of human RA is undoubtedly more complex than animal models of arthritis. Thus, strategies that inhibit multiple inflammatory chemokines, rather than a single chemokine alone, might also be a more promising direction for new RA therapies.
Vasculitis is an autoimmune disease characterized by the presence of inflammatory leukocytes in vessel walls associated with destructive damage to vessel structures. The affected vessels vary in size, type, and location according to the type of vasculitic disease; (i) small vessels, e.g., ANCA-associated vasculitis (AAV); (ii) medium vessels, e.g., Kawasaki disease (KD); and (iii) large vessels, e.g., Takayasu disease (TD) and giant cell arteritis (GCA) (58). Corticosteroids and immunosuppressive agents, such as cyclophosphamide, methotrexate and tacrolimus, are commonly used for the treatment of vasculitis. However, in some cases, the disease is refractory to these treatments, and immunosuppression often leads to significant clinical complications. Therefore, there is a need for new therapies for vasculitis that are safer and more effective than currently used treatments.
Several experimental models of vasculitis have been developed, including Polyoma virus infection-induced vasculitis, ANCA-induced vasculitis and Lactobacillus casei-induced vasculitis, but the ability to induce vasculitis in these models is limited depending on the genetic background of the mouse strain (59). Injection of a Candida albicans water-soluble fraction (CAWS) into mice leads to inflammation of the aortic root and coronary arteries and can be used as a model for KD. CAWS-induced vasculitis can be induced in a variety of genetic backgrounds and is, therefore, more versatile compared to other animal models of studying the pathogenesis of vasculitis (59), (60). In CAWS-induced vasculitis, multiple immune cells such as neutrophils, macrophages and T cells, infiltrated the inflamed cites. Additionally, neutrophils might be a key driver of inflammation in CAWS-induced vasculitis (60).
Several chemokines are elevated in the plasma and serum of vasculitis patients and are correlated with disease activity (AAV: CCL17 (61), CCL18 (62), CCL20 (63), CXCL8-11 (4), CX3CL1 (64), and XCL1 (65); TD: CCL2 (66) and CCL5 (67); GCA: CCL2 (67), CXCL9-11 (68) and CX3CL1 (69); and KD: CCL17 (70) and CXCL9/10 (70), (71)). In addition, CCL2, CCL7, CXCL2-3 and CXCL9-10 are highly expressed in the aortic root and coronary arteries of the CAWS-induced vasculitis mouse model (72), (73). Thus, the chemokine system also contributes to the development of vasculitis and RA.
In GCA, tissue-resident DCs in the adventitia of affected arteries may be a major chemokine producer in the early phase of disease onset, whereas vascular smooth muscle cells and inflammatory monocytes recruited into the artery generate chemokines during later phases (Figure 2) (74). More recently, we have demonstrated that Dectin-2-mediated CCL2 produced by tissue-resident macrophages in the aortic root and coronary arteries may be the key initiator of vascular inflammation in the CAWS-induced vasculitis. Thus, tissue-resident immune cells, such as tissue-resident macrophages and DCs, might be the main source of chemokines igniting vascular inflammation in the development of GCA and KD. However, the main sources and pathogenic role of chemokine system in other forms of human vasculitis and animal models are still unknown.
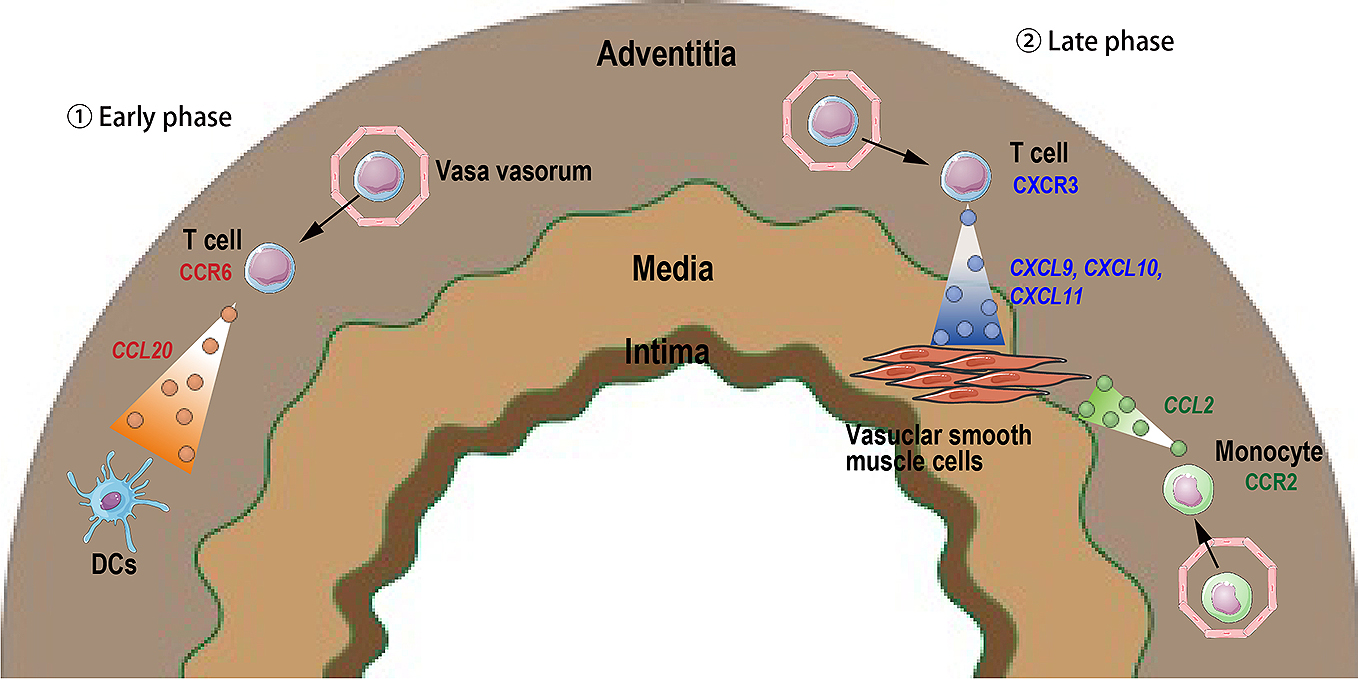
CCR2 and CX3CR1 expression on monocytes and CXCR3 and CCR6 expression on CD8+ T cells, along with high levels of the ligands for these receptors, can be observed in temporal arterial biopsies derived from patients with active GCA (Figure 2) (68), (69). In addition, expression of CXCR2 and CCR2 in the aortic root and coronary arteries is increased in CAWS-induced vasculitis mice, suggesting that CXCR2 promotes neutrophil recruitment and CCR2 controls monocyte infiltration. However, the patterns of chemokine receptor expression and their pathogenic role in human and animal models of vasculitis have not been well studied yet.
CCR2-deficient mice are resistant to the development of CAWS-induced vasculitis, suggesting that chemokines and their receptors might be attractive new targets for vasculitis treatments (75). However, the effect of inhibiting chemokines and chemokine receptors in human vasculitis and in animal models has not been well studied yet. Recently, a complementary component C5a that is a chemoattractant, but not a chemokine, can be a new promising target for vasculitis. In fact, the C5a receptor inhibitor looks very promising in the clinical trial of ANCA-associated vasculitis (76). Thus, additional studies are required to establish a new vasculitis therapy for targeting the chemokine system.
Many chemokines and their receptors have been implicated in leukocyte recruitment into inflamed tissue in RA and vasculitis. Thus, chemokines and their receptors represent promising targets for therapeutic intervention in the treatment of RA and vasculitis. In fact, inhibition of chemokines and their receptors ameliorates inflammation in animal models. However, clinical trials with chemokine and chemokine receptor blockers in human RA have failed to show clinical effects. In addition, the mechanism of action for many chemokines and their receptors in RA and vasculitis is still unknown. An important observation that requires further understanding is that different chemokines may be important at different stages of disease depending on RA and vasculitis. Recent advances in imaging technology have provided unprecedented views into immune cell function in live animals, providing an entirely new paradigm for immune cell function. In fact, our recent work using multiphoton intravital microscopy to study neutrophil entry into the joint in the KxB/N model demonstrated that the classical C5a receptor C5aR1 and the atypical C5a receptor C5aR2 were required for initial integrin-dependent neutrophil arrest on the joint endothelium but were not involved in inducing neutrophil extravasation (16), (42). By contrast, the classical chemokine receptor CXCR2 and the atypical chemokine receptor ACKR1 were involved in neutrophil diapedesis into the joint space (16), (42). In addition, other work has indicated that blockade of multiple chemokines might be more effective than single chemokine inhibition, because functions of some chemokines in immune cell trafficking overlap (57). These studies point to a need to more fully dissect the functional roles of chemokines and their receptors in the pathogenesis of RA and vasculitis. A significant need remains for the treatment of RA and vasculitis. Recent clinical studies suggest that the development of more effective inhibitors of chemokines and their receptors has untapped therapeutic potential.
This article is based on the study, which received the Medical Research Encouragement Prize of The Japan Medical Association in 2019.
None
This work of Y.M. was supported by the Japanese Society for the Promotion of Science (JSPS) Kakenhi grant number JP19K08895, AMED under Grant Number JPjm0210069h, the Takeda Science Foundation, the Maruyama Memorial Research Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, and the Medical Research Encouragement Prize of The Japan Medical Association. The work of A.D.L. was supported by grants from the National Institutes of Health and the Rheumatology Research Foundation.
Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41(5):694-707.
Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659-702.
Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12(1):5-13.
Berti A, Warner R, Johnson K, et al. Brief report: circulating cytokine profiles and antineutrophil cytoplasmic antibody specificity in patients with antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. 2018;70(7):1114-21.
Miyabe Y, Lian J, Miyabe C, et al. Chemokines in rheumatic diseases: pathogenic role and therapeutic implications. Nat Rev Rheumatol. 2019;15(12):731-46.
Komano Y, Tanaka M, Nanki T, et al. Incidence and risk factors for serious infection in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitors: a report from the Registry of Japanese Rheumatoid Arthritis Patients for Longterm Safety. J Rheumatol. 2011;38(7).
Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. 2008;Chapter(15):Unit 15.22.
Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2(5):1269-75.
Khachigian LM. Collagen antibody-induced arthritis. Nat Protoc. 2006;1(5):2512-6.
Nanki T, Shimaoka T, Hayashida K, et al. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. 2005;52(10):3004-14.
Meeuwisse CM, van der Linden MP, Rullmann TA, et al. Identification of CXCL13 as a marker for rheumatoid arthritis outcome using an in silico model of the rheumatic joint. Arthritis Rheum. 2011;63(5):1265-73.
Endo H, Akahoshi T, Takagishi K, et al. Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine Cytokine Res. 1991;10(4):245-52.
Watanabe K, Penfold ME, Matsuda A, et al. Pathogenic role of CXCR7 in rheumatoid arthritis. Arthritis Rheum. 2010;62(11):3211-20.
Yellin M, Paliienko I, Balanescu A, et al. A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(6):1730-9.
Nanki T, Imai T, Nagasaka K, et al. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46(11):2878-83.
Miyabe Y, Miyabe C, Murooka TT, et al. Complement C5a receptor is the key initiator of neutrophil adhesion igniting immune complex-induced arthritis. Sci Immunol. 2017;2(7).
Lee JH, Kim B, Jin WJ, et al. Pathogenic roles of CXCL10 signaling through CXCR3 and TLR4 in macrophages and T cells: relevance for arthritis. Arthritis Res Ther. 2017;19(1):163.
Armas-González E, Domínguez-Luis MJ, Díaz-Martín A, et al. Role of CXCL13 and CCL20 in the recruitment of B cells to inflammatory foci in chronic arthritis. Arthritis Res Ther. 2018;20(1):114.
Moschovakis GL, Bubke A, Friedrichsen M, et al. T cell specific Cxcr5 deficiency prevents rheumatoid arthritis. Sci Rep. 2017;7(1):8933.
Haringman JJ, Ludikhuize J, Tak PP. Chemokines in joint disease: the key to inflammation? Ann Rheum Dis. 2004;63(10):1186-94.
Barnes DA, Tse J, Kaufhold M, et al. Polyclonal antibody directed against human RANTES ameliorates disease in the Lewis rat adjuvant-induced arthritis model. J Clin Invest. 1998;101(12):2910-9.
Yokoyama W, Kohsaka H, Kaneko K, et al. Abrogation of CC chemokine receptor 9 ameliorates collagen-induced arthritis of mice. Arthritis Res Ther. 2014;16(5):445.
Nanki T, Takada K, Komano Y, et al. Chemokine receptor expression and functional effects of chemokines on B cells: implication in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2009;11(5):R149.
Nanki T, Urasaki Y, Imai T, et al. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J Immunol. 2004;173(11):7010-6.
Pandya JM, Lundell AC, Andersson K, et al. Blood chemokine profile in untreated early rheumatoid arthritis: CXCL10 as a disease activity marker. Arthritis Res Ther. 2017;19(1):20.
Chou RC, Kim ND, Sadik CD, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33(2):266-78.
Isozaki T, Arbab AS, Haas CS, et al. Evidence that CXCL16 is a potent mediator of angiogenesis and is involved in endothelial progenitor cell chemotaxis: studies in mice with K/BxN serum-induced arthritis. Arthritis Rheum. 2013;65(7):1736-46.
Zheng B, Ozen Z, Zhang X, et al. CXCL13 neutralization reduces the severity of collagen-induced arthritis. Arthritis Rheum. 2005;52(2):620-6.
Jacobs JP, Ortiz-Lopez A, Campbell JJ, et al. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 2010;62(7):1921-32.
García-Vicuña R, Gómez-Gaviro MV, Domínguez-Luis MJ, et al. CC and CXC chemokine receptors mediate migration, proliferation, and matrix metalloproteinase production by fibroblast-like synoviocytes from rheumatoid arthritis patients. Arthritis Rheum. 2004;50(12):3866-77.
Takayasu A, Miyabe Y, Yokoyama W, et al. CCL18 activates fibroblast-like synoviocytes in patients with rheumatoid arthritis. J Rheumatol. 2013;40(6):1026-8.
Pickens SR, Chamberlain ND, Volin MV, et al. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011;63(4):914-22.
Li JL, Lim CH, Tay FW, et al. Neutrophils self-regulate immune complex-mediated cutaneous inflammation through CXCL2. J Invest Dermatol. 2016;136(2):416-24.
Patterson AM, Schmutz C, Davis S, et al. Differential binding of chemokines to macrophages and neutrophils in the human inflamed synovium. Arthritis Res. 2002;4(3):209-14.
Ruth JH, Rottman JB, Katschke KJ, et al. Selective lymphocyte chemokine receptor expression in the rheumatoid joint. Arthritis Rheum. 2001;44(12):2750-60.
Wengner AM, Höpken UE, Petrow PK, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56(10):3271-83.
Pickens SR, Chamberlain ND, Volin MV, et al. Role of the CCL21 and CCR7 pathways in rheumatoid arthritis angiogenesis. Arthritis Rheum. 2012;64(8):2471-81.
Chen Z, Kim SJ, Essani AB, et al. Characterising the expression and function of CCL28 and its corresponding receptor, CCR10, in RA pathogenesis. Ann Rheum Dis. 2015;74(10):1898-906.
Smith E, McGettrick HM, Stone MA, et al. Duffy antigen receptor for chemokines and CXCL5 are essential for the recruitment of neutrophils in a multicellular model of rheumatoid arthritis synovium. Arthritis Rheum. 2008;58(7):1968-73.
Patterson AM, Siddall H, Chamberlain G, et al. Expression of the Duffy antigen/receptor for chemokines (DARC) by the inflamed synovial endothelium. J Pathol. 2002;197(1):108-16.
Baldwin HM, Singh MD, Codullo V, et al. Elevated ACKR2 expression is a common feature of inflammatory arthropathies. Rheumatol Oxf Engl. 2017;56(9):1607-17.
Miyabe Y, Miyabe C, Mani V, et al. Atypical complement receptor C5aR2 transports C5a to initiate neutrophil adhesion and inflammation. Sci Immunol. 2019;4(35).
Kim B, Lee JH, Jin WJ, et al. JN-2, a C-X-C motif chemokine receptor 3 antagonist, ameliorates arthritis progression in an animal model. Eur J Pharmacol. 2018;823:1-10.
Talbot J, Bianchini FJ, Nascimento DC, et al. CCR2 expression in neutrophils plays a critical role in their migration into the joints in rheumatoid arthritis. Arthritis Rheumatol. 2015;67(7):1751-9.
Amat M, Benjamim CF, Williams LM, et al. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br J Pharmacol. 2006;149(6):666-75.
Brodmerkel CM, Huber R, Covington M, et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J Immunol. 2005;175(8):5370-8.
Vierboom MP, Zavodny PJ, Chou CC, et al. Inhibition of the development of collagen-induced arthritis in rhesus monkeys by a small molecular weight antagonist of CCR5. Arthritis Rheum. 2005;52(2):627-36.
Bonelli M, Puchner A, Göschl L, et al. CCR6 controls autoimmune but not innate immunity-driven experimental arthritis. J Cell Mol Med. 2018;22(11):5278-85.
Moschovakis GL, Bubke A, Friedrichsen M, et al. The chemokine receptor CCR7 is a promising target for rheumatoid arthritis therapy. Cell Mol Immunol. 2019;16(10):791-9.
Min SH, Wang Y, Gonsiorek W, et al. Pharmacological targeting reveals distinct roles for CXCR2/CXCR1 and CCR2 in a mouse model of arthritis. Biochem Biophys Res Commun. 2010;391(1):1080-6.
Slauenwhite D, Gebremeskel S, Doucette CD, et al. Regulation of cytokine polarization and T cell recruitment to inflamed paws in mouse collagen-induced arthritis by the chemokine receptor CXCR6. Arthritis Rheumatol. 2014;66(11):3001-12.
Matsumoto N, Kon S, Nakatsuru T, et al. A novel alpha 9 integrin ligand, XCL1/lymphotactin, is involved in the development of murine models of autoimmune diseases. J Immunol. 2017;199(1):82-90.
Klimatcheva E, Pandina T, Reilly C, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16:6.
Ogata H, Takeya M, Yoshimura T, et al. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol. 1997;182(1):106-14.
Tak PP, Balanescu A, Tseluyko V, et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann Rheum Dis. 2013;72(3):337-44.
Dairaghi DJ, Zhang P, Wang Y, et al. Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin Pharmacol Ther. 2011;89(5):726-34.
Angelini A, Miyabe Y, Newsted D, et al. Directed evolution of broadly crossreactive chemokine-blocking antibodies efficacious in arthritis. Nat Commun. 2018;9(1):1461.
Watts RA, Scott DG. Recent developments in the classification and assessment of vasculitis. Best Pract Res Clin Rheumatol. 2009;23(3):429-43.
Mogi M, Liu S. Animal models of vasculitis. Methods Mol Biol. 2018;1868:223-32.
Miyabe C, Miyabe Y, Miura NN, et al. Am80, a retinoic acid receptor agonist, ameliorates murine vasculitis through the suppression of neutrophil migration and activation. Arthritis Rheum. 2013;65(2):503-12.
Dallos T, Heiland GR, Strehl J, et al. CCL17/thymus and activation-related chemokine in Churg-Strauss syndrome. Arthritis Rheum. 2010;62(11):3496-503.
Brix SR, Stege G, Disteldorf E, et al. CC chemokine ligand 18 in ANCA-associated crescentic GN. J Am Soc Nephrol. 2015;26(9):2105-17.
Eriksson P, Andersson C, Cassel P, et al. Increase in Th17-associated CCL20 and decrease in Th2-associated CCL22 plasma chemokines in active ANCA-associated vasculitis. Scand J Rheumatol. 2015;44(1):80-3.
Matsunawa M, Odai T, Wakabayashi K, et al. Elevated serum levels of soluble CX3CL1 in patients with microscopic polyangiitis. Clin Exp Rheumatol. 2009;27(1):72-8.
Blaschke S, Brandt P, Wessels JT, et al. Expression and function of the C-class chemokine lymphotactin (XCL1) in Wegener’s granulomatosis. J Rheumatol. 2009;36(11):2491-500.
Savioli B, Abdulahad WH, Brouwer E, et al. Are cytokines and chemokines suitable biomarkers for Takayasu arteritis? Autoimmun Rev. 2017;16(10):1071-8.
Dhawan V, Mahajan N, Jain S. Role of C-C chemokines in Takayasu’s arteritis disease. Int J Cardiol. 2006;112(1):105-11.
Samson M, Ly KH, Tournier B, et al. Involvement and prognosis value of CD8(+) T cells in giant cell arteritis. J Autoimmun. 2016;72:73-83.
van Sleen Y, Wang Q, van der Geest KSM, et al. Involvement of monocyte subsets in the immunopathology of giant cell arteritis. Sci Rep. 2017;7(1):6553.
Feng S, Yadav SK, Gao F, et al. Plasma levels of monokine induced by interferon-gamma/chemokine (C-X-X motif) ligand 9, thymus and activation-regulated chemokine/chemokine (C-C motif) ligand 17 in children with Kawasaki disease. BMC Pediatr. 2015;15:109.
Ko TM, Kuo HC, Chang JS, et al. CXCL10/IP-10 is a biomarker and mediator for Kawasaki disease. Circ Res. 2015;116(5):876-83.
Stock AT, Hansen JA, Sleeman MA, et al. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. 2016;213(10):1983-98.
Suzuki C, Nakamura A, Miura N, et al. Non-receptor type, proline-rich protein tyrosine kinase 2 (Pyk2) is a possible therapeutic target for Kawasaki disease. Clin Immunol. 2017;179:17-24.
Samson M, Corbera-Bellalta M, Audia S, et al. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun Rev. 2017;16(8):833-44.
Martinez HG, Quinones MP, Jimenez F, et al. Important role of CCR2 in a murine model of coronary vasculitis. BMC Immunol. 2012;13:56.
Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor Avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017;28(9):2756-67.
