Corresponding author: Masahide Takahashi, mtakaha@med.nagoya-u.ac.jp, masahide.takahashi@fujita-hu.ac.jp
DOI: 10.31662/jmaj.2020-0021
Received: March 31, 2020
Accepted: April 23, 2020
Advance Publication: July 7, 2020
Published: July 15, 2020
Cite this article as:
Takahashi M, Kawai K, Asai N. Roles of the RET Proto-oncogene in Cancer and Development. JMA J. 2020;3(3):175-181.
RET (REarranged during Transfection) is activated by DNA rearrangement of the 3′ fragment of the receptor tyrosine kinase gene, namely, RET proto-oncogene, with the 5′ fragment of various genes with putative dimerization domains, such as a coiled coil domain, that are necessary for constitutive activation. RET rearrangements have been detected in a variety of human cancers, including thyroid, lung, colorectal, breast, and salivary gland cancers. Moreover, point mutations in RET are responsible for multiple endocrine neoplasia types 2A and 2B, which can develop into medullary thyroid cancer and pheochromocytoma. Substantial effort is currently being exerted in developing RET kinase inhibitors. RET is also responsible for Hirschsprung’s disease, a developmental abnormality in the enteric nervous system. Gene knockout studies have demonstrated that RET plays essential roles in the development of the enteric nervous system and kidney as well as in spermatogenesis. Studies regarding RET continue to provide fascinating challenges in the fields of cancer research, neuroscience, and developmental biology.
Key words: RET, oncogene, DNA rearrangement, Thyroid Cancer, Lung Cancer, Multiple Endocrine Neoplasia type 2, Hirschsprung’s Disease, Development
The RET oncogene was first identified from the transfection of NIH3T3 cells in 1985 (1). RET proto-oncogene encodes a receptor tyrosine kinase and is primarily expressed as two isoforms of 1072 (short isoform) and 1114 (long isoform) amino acids by alternative splicing in the 3′ region (2). Overall, 51 carboxyl-terminal amino acids in the long isoform are replaced by nine unrelated amino acids in the short isoform. The presence of four repeated cadherin-like domains in the RET extracellular domain is unique among receptor tyrosine kinases (Figure 1) (3). As observed for cadherins, the binding of Ca2+ ions in the cadherin-like domain is necessary for RET activation (4). The extracellular domain also contains a cysteine-rich region with 16 cysteine residues (5), six of which are mutated in MEN2A families.
Analyses using in situ hybridization and immunohistochemistry have identified RET mRNA and protein expression in restricted tissues and cells during embryogenesis and after birth (6), (7). During embryogenesis, RET expression is detected in the excretory system, including the nephric duct, ureteric bud, and collecting ducts of the kidney. In the developing peripheral nervous systems, it is highly expressed in enteric neural crest cells and autonomic and dorsal root ganglia. In the central nervous system, RET protein is expressed in the neuroepithelial cells of the ventral neural tube as well as in several cranial ganglia, including facial, glossopharyngeal, trigeminal, and vagus cranial ganglia. After birth, neurons in the nervous systems mentioned above continue to express RET protein, whereas its expression has not been observed in the kidneys of adult rats.
In 1996, although GDNF does not bind to RET directly, it was found that glial cell line-derived neurotrophic factor (GDNF) could activate RET kinase. A glycosylphosphatidylinositol-anchored cell surface protein, GDNF family receptor α1 (GFR α1), is necessary for GDNF binding, and the GDNF-GFR α1 complex mediates RET dimerization (8), (9). Subsequently, three further GDNF family ligands (GFLs), including neurturin (NRTN), artemin (ARTN), and persephin, were identified and found to bind to GFR α1, α2, and α3, respectively, leading to RET activation (10), (11). GFRα family members contain three globular cysteine-rich domains (D1, D2, and D3) except for GFRα4, which lacks the N-terminal D1. GDNF functions as a homodimer and binds to the D2 domain of GFRα1(11). The structure of the GFL-GFRα complex is common among the four GFL-GFRα pairs. When two molecules of RET are recruited to the lipid raft by the GDNF-GFRα1 dimeric complex, the two cysteine-rich domains of RET are brought into close proximity, thus promoting dimer formation and the activation of tyrosine kinase (11). Recent cryo-electronic microscopic analysis has determined the structure of the NRTN-GFRα2-RET complex (12). The reconstitution of the extracellular areas of the NRTN- and GFRα2-bound RET signaling complex revealed that the RET extracellular domain interacts with the NRTN-GFRα2 complex via a large surface area. During this interaction, RET cadherin-like domains 1–3 interact with the GFRα2 D2 and D3 domains, and the unmodeled RET cysteine-rich domain is in contact with NRTN and GFRα2. This explains how complex formation leads to RET receptor dimerization. Despite large interaction surfaces, the GFRα2 and RET extracellular domains do not appear to be associated in the absence of NRTN.
Gene knockout (KO) mice for GDNF, GFRα1, or RET die soon after birth and share morphologically similar phenotypes, including a lack of enteric neurons in the entire enteric nervous system, kidney agenesis or severe dysgenesis, and the impairment of spermatogenesis(13), (14), (15), (16). Sensory, sympathetic, parasympathetic, and motor neurons are affected in these mice to varying degrees. Moreover, mice with an introduced mutation at tyrosine 1062 in RET, whose phosphorylation by GDNF stimulation is crucial for RET downstream signaling, such as the RAS-MAPK and PI3K-AKT pathways, showed phenotypes similar to, but milder than, RET KO mice (17), (18). These findings confirm the biological importance of GDNF/GFRα1/RET complex formation and resultant signaling activation during morphogenesis.
Similarly, mice that lack NRTN or GFRα2 have similar defects in enteric and parasympathetic innervations (19), (20). The parasympathetic cholinergic innervation is dramatically reduced in the lacrimal and submandibular salivary glands as well as in the intestines. However, NRTN-deficient mice grow normally, whereas GFRα2-deficient mice show growth retardation. GFRα3 KO mice exhibit severe defects in the superior cervical ganglion but not in other ganglia (21). Therefore, each GFL-GFRα-RET complex has been found to have specific roles in vivo.
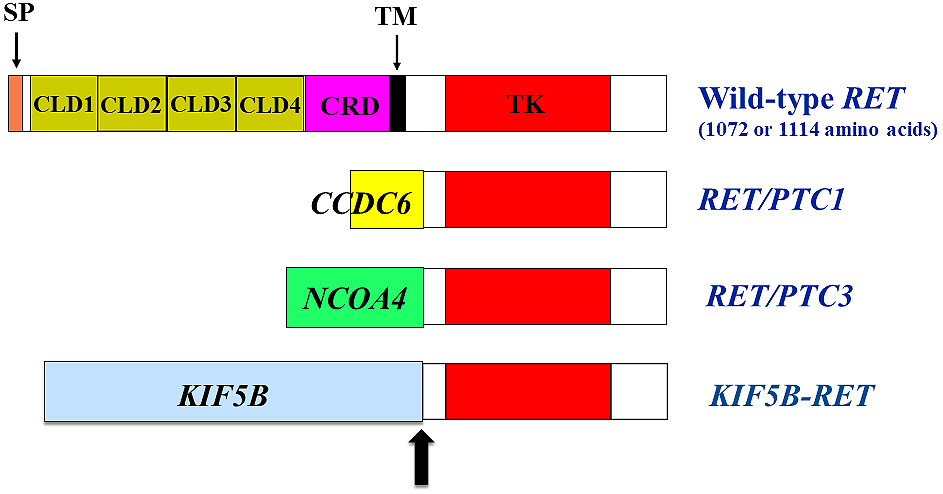
To date, RET fusion with other partner genes has been reported in a variety of human cancers, including papillary thyroid carcinoma (PTC) (22), (23), (24) and non-small cell lung cancers (NSCLCs) (25), (26), (27). RET fusion has been detected in 5%–35% of adult PTCs, in which rearrangement with the CCDC6 gene has most frequently been observed (named RET/PTC1, Figure 1). RET and CCDC6 genes are located on the long arm of chromosome 10, and this gene fusion is induced by intrachromosomal inversion. The other 5′ partner genes for RET fusion in PTC include PRKAR1A, NCOA4, GOLGA5, TRIM24, TRIM33, KTN1, and RFG9 (24). These genes, except for NCOA4, which is located on chromosome 10, generate fusions with RET by interchromosomal translocation. After the Chernobyl nuclear reactor accident occurred in 1986, the occurrence of childhood PTC in contaminated regions dramatically increased, reaching approximately 10-fold increase 10 years after the accident. Of the childhood cases, ~60% to ~70% displayed RET rearrangement with 5′ fragment of the NCOA4 gene (named RET/PTC3, Figure 1), and this fusion, like the CCDC6-RET fusion, was induced by the paracentric inversion of chromosome 10 (23), (28).
Breakpoints preferentially occur in restricted regions of RET, most commonly within intron 11(24). This results in fusions, including in the cytoplasmic kinase domain of RET. Less frequent breakpoints involve introns 7 and 10, leading to the inclusion of the RET transmembrane domain. The 5′ partner genes contain distinct putative dimerization domains, such as a coiled coil domain, which mediates the ligand-independent activation (dimerization) of chimeric RET oncoproteins.
RET fusion has also been identified in 1%–2% of NSCLCs (25), (26), (27), with KIF5B-RET being the most commonly identified (Figure 1). CCDC6, NCOA4, and TRIM33 are also partner 5′ genes for RET fusion in NSCLCs (24). KIF5B is located on the short arm of chromosome 10, and KIF5B-RET fusion is created via pericentric inversion.
The next-generation DNA and/or RNA sequencing approach has led to the identification of RET fusion in a variety of cancers, including colorectal, breast, ovarian, and salivary gland cancers, as well as Spitz tumors and chronic myeloproliferative neoplasms (29), (30), (31). Large-scale analyses have reported that RET fusion can be detected in 0.2% of colorectal cancers (6/3117 cases) (32) and 0.1% of breast cancers (8/9693 cases) (33). CCDC6-RET and NCOA4-RET fusions have been identified in both cancers. Interestingly, a high frequency of RET fusion (42.4%), including NCOA4-RET and TRIM27-RET, has been detected in salivary intraductal carcinomas, suggesting a role of RET fusion in the development of a particular type of salivary gland cancer (34).
Germline RET mutations are responsible for the development of autosomal dominant multiple endocrine neoplasia types 2A and 2B (MEN2A and MEN2B) and familial medullary thyroid carcinoma (FMTC) (35), (36), (37), all of which confer an increased risk (~100%) of developing medullary thyroid carcinoma (MTC). MEN2A is characterized by the presence of MTC combined with the development of pheochromocytoma and parathyroid hyperplasia/adenoma in ~50% and ~20% of affected family members, respectively. Moreover, lichen amyloidosis is occasionally observed. MEN2B is characterized by the development of MTC with an early age of tumor onset and displays a more complex phenotype, including pheochromocytoma, mucosal neuroma, ganglioneuromatosis of the intestine, thickening of corneal nerves, and marfanoid habitus. FMTC only develops MTC in families, usually in the later stage of life.
MEN2A mutations in the majority of families (~95%) have been identified in one of six cysteine residues (codons 609, 611, 618, and 620 in exon 10, and codons 630 and 634 in exon 11) in the cysteine-rich region of the RET extracellular domain (Figure 2). Of these cysteine mutations, missense mutations at codon 634 have been identified in ~85% of the affected MEN2A family members (38). We and Santoro et al. previously elucidated the molecular mechanism of RET activation by cysteine mutations. When a cysteine residue is substituted with a non-cysteine residue, a partner cysteine that is normally involved in the formation of an intramolecular disulfide bond is freed and forms an aberrant intermolecular covalent disulfide bond between two mutant RET, leading to its dimerization and activation (39), (40).
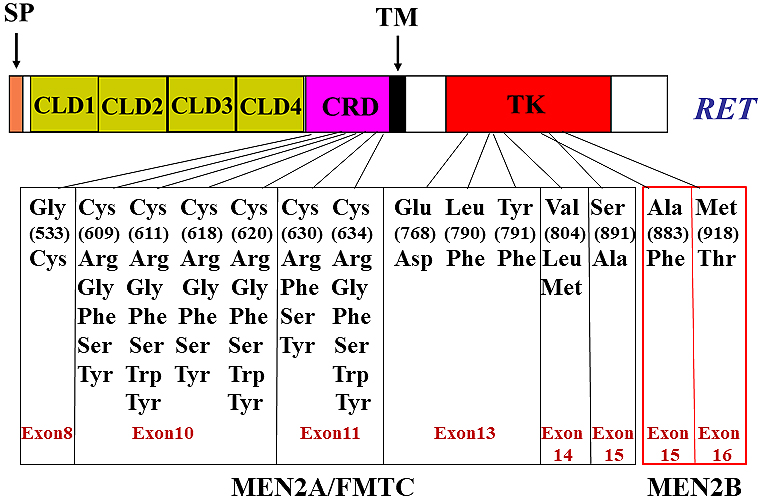
Cysteine mutations in the RET extracellular domain also cause the FMTC phenotype. Interestingly, contrary to MEN2A families, a higher frequency (~60%) of non-cysteine 634 substitutions (cysteines 609, 611, 618, 620, and 630) and a lower frequency (~30%) of cysteine 634 substitutions have been detected in FMTC families (41). We found that the transforming activities of RET with cysteine 609, 611, 618, or 620 mutations were considerably lower than those of RET with a cysteine 634 mutation because of the impairment of cell surface expression of the former four cysteine mutants (42). The low transforming activity may predispose to the development of FMTC rather than MEN2A. Moreover, Gly533Cys (G533C) (exon 8 in the RET extracellular domain) (43), Glu768Asp (E768D), Leu790Phe (L790F), Tyr791Phe (Y791F), Val804Met/Leu (V804M/L), and Ser891Ala (S891A) substitution (exons 13–15 in the RET kinase domain) (24) have been reported in some families of MEN2A and/or FMTC (Figure 2).
Two missense mutations, Met918Thr (M918T) (exon 16) and Ala883Phe (A883F) (exon 15), in the kinase domain are associated with MEN2B (Figure 2). Of the MEN2B patients, ~95% carry the M918T mutation and fewer than 4% carry the A883F mutation (44). The M918T mutation increases the kinase activity as monomers and the presentation of substrates for trans-autophosphorylation (40), which is conferred by conformational changes in the activation loop of the kinase domain. However, the mechanisms by which the different clinical phenotypes between MEN2A and MEN2B develop remain elusive.
According to data published in a public database in 2015 (Catalog of Somatic Mutations in Cancer), somatic RET mutations have been detected in 677 of 1662 (41%) samples from patients with MTC. M918T mutations were most frequent in these patients, and cysteine mutations (C634, C630, C618, and C611) as well as E768D, V804M/L, and A883F mutations were observed in sporadic MTCs (24). Oncogenic RET mutations were also detected in 0.5% (8/1489) of colorectal cancer and 0.2% (16/9693) of breast cancer, including Glu511Lys (E511K), G533C, Cys634Arg (C634R), E768D, V804M, and M918T mutations (32), (33), (45). Moreover, the increasing use of NGS has uncovered RET mutations in a variety of cancers, including endometrial and ovarian cancers, hepatomas, skin melanomas, glioblastoma multiforme, meningiomas, gastrointestinal stromal tumors, Merkel cell carcinomas, paragangliomas, and atypical lung carcinoids (29).
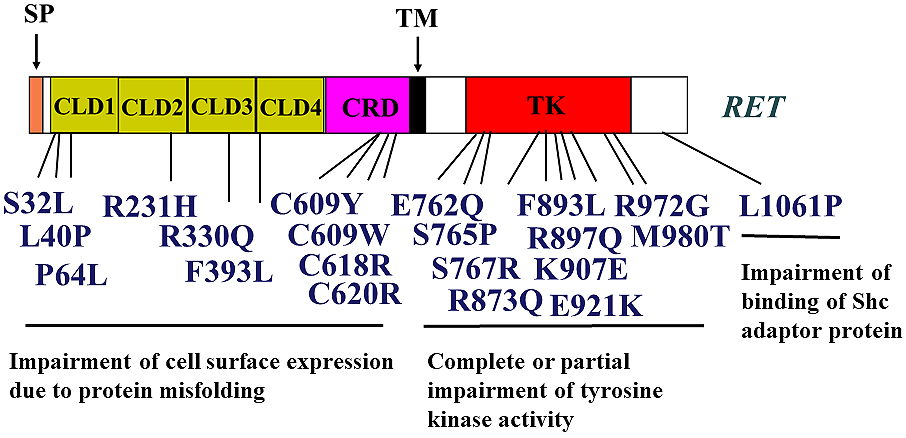
Hirschsprung’s disease (HSCR) is a congenital malformation associated with aganglionosis of the gastrointestinal tract. RET mutations account for ~50% of familial and 10%–20% of sporadic cases of HSCR (46), (47), (48), (49). A variety of missense, nonsense, and frameshift mutations have been identified along the entire coding sequence of RET (Figure 3). Moreover, partial or complete deletion of one allele of RET has been reported in some HSCR cases. Furthermore, mutations in non-coding sequences, including the promoter/enhancer regions, have been detected in HSCR, resulting in the alteration of the regulation of RET expression (50). Interestingly, the penetrance of the mutant allele in familial HSCR is low and is significantly higher in males than in females, suggesting the existence of one or more modifier genes in HSCR.
We and others have performed functional analyses of missense mutations in the RET coding region identified in patients with HSCR. Based on these studies, four mechanisms appear to be responsible for HSCR development (Figure 3). (1) Most mutations in the RET extracellular domain impair its cell surface expression, most likely because of misfolding of the RET protein (42), (51), (52), (53). Low levels of RET on the cell surface cannot transmit sufficient signals to elicit GDNF-mediated biological activities in enteric neuroblasts during embryogenesis. (2) Mutations in the kinase domain that occur at highly conserved amino acids among receptor tyrosine kinase families almost completely abolish RET kinase activity (54), (55), (56). (3) Mutations in the kinase domain (e.g., Glu762Gln (E762Q), Ser767Arg (S767R), Arg972Gly (R972G), and Met980Thr (M980T)) markedly impair the activity of PLCγ, whereas RAS and PI3K pathways are mildly affected (56). (4) Mutations in the carboxyl-terminal tail impair intracellular signaling because of decreased SHC binding to RET (57), (58), (59).
Moreover, RET mutations are less commonly associated with congenital abnormalities of the kidney and urinary tract and congenital central hypoventilation syndrome (CCHS), also known as “Ondine’s Curse” (60). In some cases, the CCHS presents together with the HSCR.
A variety of multikinase inhibitors with activity against RET have been developed and are available to treat RET-altered cancers. These include cabozantinib, vandetanib, lenvatinib, ponatinib, sorafenib, sunitinib, alectinib, regorafenib, and RXDX-105, which are classified into distinct groups by their mode of kinase inhibition (60). For example, vandetanib and sunitinib are type I inhibitors that bind within the adenosine 5′-triphosphate (ATP)-binding pocket in the active conformation of the RET kinase (61). Cabozantinib, sorafenib, ponatinib, and RXDX-105 are type II inhibitors that bind within the ATP-binding pocket in the inactive conformation of the RET kinase (61), (62), (63). However, the clinical efficacy of multikinase inhibitors in RET-altered patients appears limited (lower overall objective response ratios (ORRs) and shorter progression-free survival) and has significant off-target toxicities (60). This is most likely because these inhibitors are not specifically designed for targeting RET, and their effects were initially focused on other kinases, including VEGFR, EGFR, MET, and ALK. Cabozantinib, vandetanib, and lenvatinib more potently inhibit VEGFR2, despite the RET kinase domain having significant homology to that of VEGFR2.
Secondary RET mutations that confer the resistance of RET-altered cancers to multikinase inhibitors during therapy have been reported. V804L and V804M are representative examples that alter the valine residue in the gatekeeper site of RET (64), (65). These mutations are thought to introduce steric clashes between the leucine and methionine side chains and the 4-bromo-2-fluorophenyl group of vandetanib. I788N is another secondary mutation known as the non-gatekeeper mutation (62).
Recently, RET-specific kinase inhibitors LOXO-292 and BLU-667 have been developed to inhibit the kinase activity of both wild-type RET and the gatekeeper mutant RET (66), (67), (68). ORR (>50%) and the duration of response (>6 months) using these inhibitors showed greater benefit for RET-altered patients in clinical trials. Intrinsic resistance to multikinase inhibitors occurs in cancers that carry the KIF5B-RET fusion gene. Notably, the intrinsic resistance conferred by this fusion has been overcome with treatment of LOXO-292 and BLU-667, stressing the necessity for the development of RET-specific inhibitors.
RET was first identified 35 years ago, and its oncogenic rearrangements and mutations have been reported in a variety of cancers, including thyroid cancer, NSCLC, MEN2A, and MEN2B. Recent developments in RET-specific kinase inhibitors are making progress in providing considerable benefit to patients with RET-altered cancer. Moreover, studies regarding GDNF-RET signaling continue to have a profound impact on neuroscience and developmental biology. Based on the efficacy of GDNF as a survival factor of dopaminergic and motor neurons, establishing neurons expressing GDNF from iPS or ES cells could be beneficial for treating neurodegenerative diseases, such as Parkinson’s disease. Future studies regarding RET functions will further contribute to a deeper understanding of the molecular mechanisms of the enteric nervous system and kidney development as well as of spermatogenesis.
This article is based on the study, which received the Medical Award of The Japan Medical Association in 2019.
None
Masahide Takahashi is one of the Consultants of JMA Journal and on the journal's Editorial Staff. He was not involved in the editorial evaluation or decision to accept this article for publication at all.
Takahashi M, Ritz J, Copper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581-8.
Tahira T, Ishizaka Y, Itoh F, et al. Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene. 1990;5(1):97-102.
Ibanez CF. Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol. 2013;5(2).
Nozaki C, Asai N, Murakami H, et al. Calcium-dependent Ret activation by GDNF and neurturin. Oncogene. 1998;16(3):293-9.
Takahashi M, Buma Y, Iwamoto T, et al. Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene. 1988;3(5):571-8.
Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119(4):1005-17.
Tsuzuki T, Takahashi M, Asai N, et al. Spatial and temporal expression of the ret proto-oncogene product in embryonic, infant adult rat tissues. Oncogene. 1995;10(1):191-8.
Jing S, Wen D, Yu Y, et al. GDNF-induced activation of the Ret protein tyrosine kinase is mediated by GDNFR-(, a novel receptor for GDNF. Cell. 1996;85(7):1113-24.
Treanor JJS, Goodman L, de Sauvage F, et al. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382(6586):80-3.
Klein RD, Sherman D, Ho W-H, et al. A GPI-linked protein that interacts with Ret to form a candidate neurturin receptor. Nature. 1997;387(6634):717-21.
Airaksinen MS, Saarma M. The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3(5):383-94.
Bigalke JM, Albara S, Roth R, et al. Cryo-EM structure of the activated RET signaling complex reveals the importance of its cysteine-rich domain. Sci Adv. 2019;5(7):eaau4202.
Schuchardt A, D’Agati V, Larsson-Blomberg L, et al. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367(6461):380-3.
Sanchez MP, Silos-Santiago I, Frisen J, et al. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382(6586):70-3.
Cacalano G, Farinas I, Wang L-C, et al. GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21(1):317-24.
Meng X, Lindahl M, Hyvonen ME, et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287(5457):1489-93.
Jijiwa M, Fukuda T, Kawai K, et al. A targeting mutation of tyrosine 1062 in Ret causes a marked decrease of enteric neurons and renal hypoplasia. Mol Cell Biol. 2004;24(18):8026-36.
Jijiwa M, Kawai K, Fukihara J, et al. GDNF-mediated signaling via RET tyrosine 1062 is essential for maintenance of spermatogonial stem cells. Genes Cells. 2008;13(4):365-74.
Heuckeroth RO, Enomoto H, Grider JR, et al. Gene targeting reveals a critical role for neurturin in the development and maintenance of enteric, sensory, and parasympathetic neurons. Neuron. 1999;22(2):253-63.
Rossi J, Luukko K, Poteryaev D, et al. Retarded growth and deficits in the enteric and parasympathetic nervous system in mice lacking GFRα2, a functional neurturin receptor. Neuron. 1999;22(2):243-52.
Nishino J, Mochida K, Ohfuji Y, et al. GFRα3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron. 1999;23(4):725-36.
Grieco M, Santoro M, Berlingieri MT, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60(4):557-63.
Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol. 2002;13(1):3-16.
Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. 2016;12(4):192-202.
Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusion in lung cancer. Nat Med. 2012;18(3):375-7.
Takeuchi K, Soda M, Togashi Y, et al. RET, ROS and ALK fusions in lung cancer. Nat Med. 2012;18(3):378-81.
Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382-4.
Ito T, Seyama T, Iwamoto KS, et al. Activated RET oncogene in thyroid cancers of children from area contaminated by Chernobyl accident. Lancet. 1994;344(8917):259.
Kato S, Subbiah V, Marchlik E, et al. RET aberration in diverse cancers: Next-generation sequencing of 4871 patients. Clin Cancer Res. 2017;23(8):1988-97.
Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
Ballerini P, Struski S, Cresson C, et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012;26(11):2384-9.
Le Rolle AF, Klempner SJ, Garrett CR, et al. Identification of characterization of RET fusion in advanced colorectal cancer. Oncotarget. 2015;6:28929-37.
Paratala BS, Chung JH, Williams CB, et al. RET rearrangement are actionable alterations in breast cancer. Nat Commun. 2018;9:4821.
Skalova A, Vanecek T, Uro-Coste E, et al. Molecular profiling of salivary gland intraductal carcinoma revealed a subset of tumors harboring NCOA4-RET and novel TRM27-RET fusions: A report of 17 cases. Am J Surg Pathol. 2018;42(11):1445-55.
Mulligan LM, Kwok JBJ, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458-60.
Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET protooncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2(7):851-6.
Hofstra RMW, Landsvater RM, Ceccherini I, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367(6461):375-6.
Mulligan LM, Eng C, Healey CS, et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet. 1994;6(1):70-4.
Asai N, Iwashita T, Matsuyama M, et al. Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations. Mol Cell Biol. 1995;15(3):1613-9.
Santoro M, Carlomagno F, Romano A, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995;267(5196):381-3.
Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. JAMA. 1996;276(19):1575-9.
Ito S, Iwashita T, Asai N, et al. Biological properties of Ret with cysteine mutations correlate with multiple endocrine neoplasia type 2A, familial medullary thyroid carcinoma, and Hirschsprung’s disease phenotype. Cancer Res. 1997;57(14):2870-2.
Castro MR, Thomas BC, Richards ML, et al. Multiple endocrine neoplasia type 2A due to an exon 8 (G533C) in a large North American kindred. Thyroid. 2013;23(12):1547-52.
Smith DP, Houghton C, Ponder BA, et al. Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene. 1997;15(10):1213-7.
Mendes Oliveira D, Grillone K, Mignogna C, et al. Next-generation sequencing analysis of receptor-type tyrosine kinase genes in surgically resected colon cancer: Identification of gain-of-function mutations in the RET proto-oncogene. J Exp Clin Cancer Res. 2018;37(1):84.
Romeo G, Ronchetto P, Luo Y, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367(6461):377-8.
Edery P, Lyonnet S, Mulligan LM, et al. Mutations of the RET protooncogene in Hirschsprung’s disease. Nature. 1994;367(6461):378-80.
Angrist M, Bolk S, Thiel B, et al. Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet. 1995;4(5):821-30.
Attie T, Pelet A, Edery P, et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet. 1995;4(8):1381-6.
Emison ES, Garcia-Barcelo M, Grice EA, et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet. 2010;87(1):60-74.
Iwashita T, Murakami H, Asai N, et al. Mechanisms of Ret dysfunction by Hirschsprung mutations affecting its extracellular domain. Hum Mol Genet. 1996;5(10):1577-80.
Carlomagno F, De Vita G, Berlingieri MT, et al. Molecular heterogeneity of RET loss of function in Hirschsprung’s disease. EMBO J. 1996;15(11):2717-25.
Cosma MP, Cardone M, Carlomagno F, et al. Mutations in the extracellular domain cause RET loss of function by a dominant negative mechanism. Mol Cell Biol. 1998;18(6):3321-9.
Pasini B, Borrello MG, Greco A, et al. Loss of function effect of RET mutations causing Hirschsprung disease. Nat Genet. 1995;10(1):35-40.
Pelet A, Geneste O, Edery P, et al. Various mechanisms cause RET-mediated signaling defects in Hirschsprung’s disease. J Clin Invest. 1998;101(6):1415-23.
Iwashita T, Kurokawa K, Qiao S, et al. Functional analysis of RET with Hirschsprung’s mutations affecting its kinase domain. Gastroenterology. 2001;121(1):24-33.
Geneste O, Bidaud C, De Vita G, et al. Two distinct mutations of the RET receptor causing Hirschsprung’s disease impair the binding of signaling effector to a multifunctional docking site. Hum Mol Genet. 1999;8(11):1989-99.
Ishiguro Y, Iwashita T, Murakami H, et al. The role of amino acids surrounding tyrosine 1062 in Ret in specific binding of the Shc phosphotyrosine-binding domain. Endocrinology. 1999;140(9):3992-8.
Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12(4):361-73.
Drilon A, Hu ZI, Lai GGY, et al. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15(3):151-67.
Plaza-Menacho I, Mologni L, McDonald NQ, et al. Mechanisms of RET signaling in cancer: current and future implications for targeted therapy. Cell Signal. 2014;26(8):1743-52.
Plenker D, Riedel M, Brägelmann J, et al. Drugging the catalytically inactive state of RET kinase in RET-rearranged tumors. Sci Transl Med. 2017;9(394):eaah6144.
Li GG, Somwar R, Joseph J, et al. Antitumor activity of RXDX-105 in multiple cancer types with RET rearrangements or mutations. Clin Cancer Res. 2017;23(12):2981-90.
Dagogo-Jack I, Stevens SE, Lin JJ, et al. Emergence of a RET V804M gatekeeper mutation during treatment with Vandetanib in RET-rearranged NSCLC. J Thorac Oncol. 2018;13(11):e226-7.
Huang Q, Schneeberger VE, Luetteke N, et al. Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol Cancer Ther. 2016;15(10):2521-9.
Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29(8):1869-76.
Subbiah V, Gainor JF, Rahal R, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018;8(7):836-49.
Subbiah V, Yang D, velcheti V, et al. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J CIin Oncol. 2020;38(11):1209-1221.